
More Information
Submitted: 18 July 2019 | Approved: 23 July 2019 | Published: 24 July 2019
How to cite this article: Alqoaer K, Asaad Z, Halabi M. Severe Infantile Transaldolase deficiency: A case report. J Adv Pediatr Child Health. 2019; 2: 001-003.
doi: 10.29328/journal.japch.1001003
Copyright License: © 2019 Alqoaer K, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Transaldolase; Pentose pathway

Severe Infantile Transaldolase deficiency: A case report
Khaled Alqoaer*, Ziad Asaad and Maisa Halabi
Department of Pediatrics, King Salman Armed Forces Hospital, Tabuk, Saudi Arabia
*Address for Correspondence: Khaled Alqoaer, Department of Pediatrics, King Salman Armed Forces Hospital, Tabuk, Saudi Arabia, Tel: 00966533033970; Email: [email protected]; [email protected]
Transaldolase TALDO deficiency is a rare autosomal recessive disorder of the pentose phosphate pathway. It has variable presentations with poor outcome when present early in life. We present a young Saudi infant with a fatal early presentation of TALD deficiency.
Transaldolase TALDO is an enzyme required in pentose phosphate pathway (PPP). It is an alternative route for glucose oxidation. It links the pentose phosphate pathway to glycolysis. The end products are D-ribose 5-phosphate and Nicotinamide adenine dinucleotidephosphate NADPH. In the absence of TALDO, toxic intermediate products may accumulate and lead to variety of clinical features. TALDO deficiency is a rare autosomal recessive disorder with main presentations: an early onset severe form that can be fatal and a late onset presentation which comprises a slowly progressive less severe form of the disease. The diagnosis can be made by special tests in urine and plasma or by enzymatic assay. However, the diagnosis nowadays is usually confirmed by DNA analysis of the TALDO1 gene. No effective treatment is available so far.
This is a four months old girl who was born at term by normal spontaneous vaginal delivery to a 39 years old mother. The parents are second degree cousins. They have had another five healthy children. The pregnancy was uneventful. The birth weight was 2.1 kg (-2.4 SD), length 48 cm (-0.5 SD) and head circumference 31 cm (-2.8 SD). The patient was admitted on the second day of life to NICU for six days because of indirect hyberbilirubenemia (total bilirubin 158 normal 5.1–20.5 mmol/L) and received phototherapy. His platelet count was 45 (normal 223–461 cells/mL). The jaundice improved and the platelet count rose to 61. The patient was discharged in a stable condition. He was seen after one week where the repeated CBC was normal and the platelet count was 200. At the age of four months the patient presented with abdominal distension that started according to the parents at the age of one moth and was gradually increasing. There was no history of vomiting, diarrhea or constipation, fever, skin rash or decreased activity. The baby was on breast feeding with acceptable intake. The physical examination revealed normal vital signs with subtle dysmorphic features (low set ears and depressed nasal bridge). The skin examination was unremarkable. The weigh at age of four months was 5.4 kg (-1.4 SD), length 53 cm (-3.7 SD) and the head circumference 35 cm (-4.7 SD). The abdomen was distended with no guarding, tenderness or dilated veins. There was huge splenomegaly up to the left iliac fossa but no hepatomegaly. The shifting dullness was positive for ascites. The rest of physical examination was unremarkable. The investigations showed persistent low white blood count as low as 3.3 (normal 7.73–13.12 cells/mL), absolute neutrophil count 860, lymphocytes count of 2060, monocytes 380, basophils and eosinophil of zero. The hemoglobin dropped to 6.1 (normal 10.4–12.5 g/dl) with normochromic, normocytic picture. The reticulocyte count 6.9%. The platelet count was persistently low. The peripheral blood film showed pancytopenia but no abnormal cells. Serum urea, creatinine and electrolytes were all normal. Liver enzymes were mildly elevated. ALT 135 (normal 3–30 U/L) and AST 81 (normal 2–40 U/L) while the serum bilirubin was initially 95 mmol/L with direct fraction of 50 mmol/L. Later on the serum bilirubin increased gradually till reached 539 mmol/L with direct fraction of 355 mmol/L. Alkaline phosphatase 1107 (normal 110–400 U/L), albumin 13 (normal 30–46 g/L) and GGT 77 (normal 12–55 U/L)). Urine analysis was negative for protein and others. The coagulation profile was initially normal but later on became prolonged (INR 2.7, PT 37 seconds and PTT 61 seconds). D-Dimer 7.36 (0-0.50 ug/ml), fibrinogen 100 (normal 250-350 mg/dl). Serum alpha-fetoprotein was elevated (>1000 ng/ml). Triglycerides, Serum ferritin, ammonia, lactate and blood gas were all normal. Serum bile acids was elevated 87 (normal <10 umol/l) and serum Lactate dehydrogenase LDH was mildly elevated (438 normal 180-430 u/l) Cytomegalo virus serology (IgM ) was positive while Ebestien bar virus serology and PCR were negative. Stool alpha-fetoprotein was normal. Thyroid profile (TSH, T3 and T4) was normal. Bone marrow aspiration showed scattered hematobiotic precursors. No increase in blast cells seen and no storage cells were identified. Urine for organic acids showed high methionine (Methionine level @ 82 uM (normal: 6-63 uM). Also the serum amino acids showed moderate hyper-methioninemia. Tandem mass spectrometry from dried blood spot showed low b-Glcocerbrocidase of 2.3 (normal > 2.5 umol/l/h) but acid Sphingomylase was normal. Molecular genetic testing for Gaucher disease showed no pathogenic mutation. Skeletal survey x-ray showed Widened metaphysis of the lower long bones only. CT brain showed no evident intra-cranial calcifications. Abdominal CT scan revealed hepatosplenomegaly and ascites with patent Portal veins and no varices. The liver was enlarged in size (span 12 cm), more of its left lobe that cross to the other side with homogenous attenuation and regular contour. The spleen was: hugely enlarged reaches The left iliac fossa (about 10 cm) with no focal lesions. Splenic vein was prominent (4.2 mm) but no gross varices noted. Moderate to marked ascites was noted. The Eye exam by ophthalmologist was unremarkable. Echocardiogram was done and it showed just a small ASD secondum. The patient was managed with frequent blood, fresh frozen plasma and platelet transfusions. Multiple infusions of human albumin were given because of progressive abdominal distension and ascites with minimal response. Intravenous immunoglobulin was given once. A sex weeks course of intravenous gancyclvir was administered as per our infectious disease team. Diuretics in form of spironolactone and furosemide in addition to vitamin k and multivitamins were added as well. The patient didn’t show any improvement. The patient condition rapidly deteriorated and went in liver and respiratory failure with uncontrollable pulmonary hemorrhage till passed away. Later on the analysis of whole exome sequence showed homozygous for TALDO1 c.793del, p.(Gln265Argfs*56), which is pathogenic and diagnostic for Transaldolase deficiency.
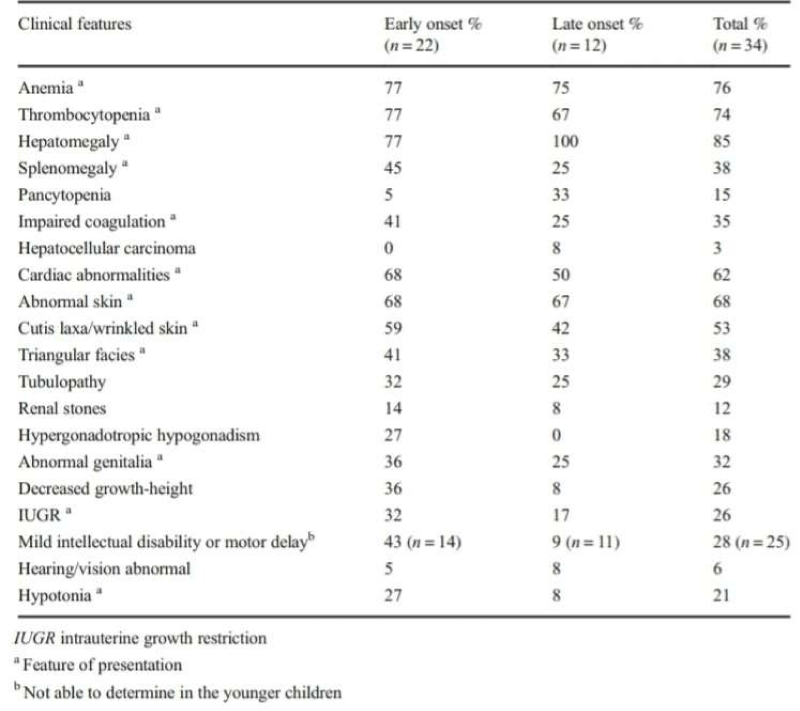
Table 1: Clinical features of Transaldolase deficiency. Williams M, et al. J Inherit Metab Dis. 2019 [8].
Pentose pathway is an alternative route for glucose oxidation. The end products of the pathway are D-ribose 5-phosphate and Nicotinamide adenine dinucleotidephosphate NADPH. Trans ketolase and transaldolase, are essential enzymes required to link the pentose phosphate pathway to glycolysis in a reversible fashion. These two enzymes convert pentose 5-phosphate into glycolytic intermediates [1,2]. By conversion of the carbon structure among different sugars, it enables the production of fructose-6 phosphate and erythrose-4-phosphate from glyceraldehyde-3-phosphate and sedoheptulose-7-phosphate. Intermediate toxic metabolites such as ribitol, D-arabitol and erythritol may accumulates In the absence of transaldolase [3]. TALDO deficiency is a rare autosomal recessive inborn error of the pentose phosphate pathway first described in 2001 [1].
To date, has been diagnosed in 41 patients worldwide. The largest number of these patient where interestingly Saudis (12 patients) [4-8]. Two main presentations of TALDO has been recognized so far: an early onset presentation (prenatal or neonatal disease) which is the severe form, in most of the cases rapidly fatal, and a late onset presentation (early or late infantile form) with slowly progressive and milder form of the disease. The liver dysfunction begins in early fetal life in both presentations. In the early onset presentation, liver involvement usually present in the form of coagulopathy, transaminitis, hypo albuminemia, and skin changes in newborns. Progressive liver failure is the main factor in the reduced lifespan. In patients with later onset of the disease, liver dysfunction is milder and progresses more slowly. Renal tubular dysfunction may occur and usually present with low molecular weight proteinuria [8]. The mechanism of liver and kidney involvement in TALDO is most likely secondary to the toxic accumulation of sugars and polyols (sedoheptulose-7-phosphate, ribose-5-phosphate, ribulose-5-phosphate, xylulose-5-phosphate and C5-polyols: D-ribitol; D-arabitol; and D-xylitol) on the hepatocytes and kidney tubules [2,9,10]. The concentrations of these toxic metabolites are notably the highest in the first weeks of life. Polyol concentrations seem to decrease with age. The other clinical features are summarized in table 1.
Our patient represented the severe neonatal type of TALDO deficiency. The interesting feature is the huge splenomegaly which was more impressive than hepatomegaly. In most of the reported cases the hepatomegaly was more impressive. The diagnosis of TALDO deficiency is challenging. As the clinical presentation varies, the diagnosis usually is confirmed by DNA analysis of the TALDO1 gene. Abnormal polyols and/or sugars (erythritol, arabitol, ribitol, sedoheptitol, perseitol, sedoheptulose, mannoheptulose, and sedoheptulose-7P) can be detected in urine. Erythritol, arabitol, and ribitol may be mildly elevated in the plasma and cerebrospinal fluid. Previously erythronic acid has been labeled as a biomarker in TALDO-D. Aminoaciduria is not uncommon but usually not specific and of the form commonly seen with liver dysfunction (high glutamine, methionine, and ethanolamine). Enzymatic analysis of TALDO enzyme can be measured and usually showed lower level than control. Most patients are homozygous for a mutation in TALDO1 in keeping with the high frequency of consanguinity. Missense mutations, in frame deletions, and frameshift mutations have been detected [8]. No Effective treatment for TALDO deficiency is available up to date. Liver transplantation has a potential risk of disease recurrence. N-acetylcysteine, decreasing oxidative stress with antioxidants (e.g., vitamins C or E), and reducing polyols or sugar-P accumulation by using specific inhibitors of the PPP were suggested [11]. Only N-acetylcysteine is shown to work on and the response was specifically reported after challenging with acetaminophen [4,5,7]. In conclusion, TALDO deficiency is a rare in born error of metabolism of the pentose phosphate pathway with multisystem variable presentations that should be considered in any pediatric patient with unexplained hepatosplenomegaly or hepatic failure. However, no effective treatment is available so far.
- Verhoeven NM, Huck JH, Roos B, Struys EA, Salomons GS, et al. Transaldolase deficiency: liver cirrhosis associated with a new inborn error in the pentose phosphate pathway. Am J Hum Genet. 2001; 68: 1086-1092. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/11283793
- Perl A. The pathogenesis of transaldolase deficiency. IUBMB Life. 2007; 59: 365-373. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/17613166
- Wamelink MM, Smith DE, Jansen EE, Verhoeven NM, Struys EA, et al. Detection of transaldolase deficiency by quantification of novel seven-carbon chain carbohydrate biomarkers in urine. J Inherit Metab Dis. 2007; 30: 735-742. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/17603756
- Eyaid W, Al Harbi T, Anazi S, Wamelink MM, Jakobs C, et al. Transaldolase deficiency: report of 12 new cases and further delineation of the phenotype. J Inherit Metab Dis. 2013; 36: 997-1004. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23315216
- Rodan LH, Berry GT. N-Acetylcysteine Therapy in an Infant with Transaldolase Deficiency Is Well Tolerated and Associated with Normalization of Alpha Fetoprotein Levels. JIMD Rep. 2017; 31: 73-77. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27130472
- Lipiński P, Pawłowska J, Stradomska T, Ciara E, Jankowska I, et al. Long-Term Systematic Monitoring of Four Polish Transaldolase Deficient Patients. JIMD Rep. 2018; 42: 79-87. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29292491
- Lee-Barber J, English TE, Britton JF, Sobreira N, Goldstein J, et al. Apparent Acetaminophen Toxicity in a Patient with Transaldolase Deficiency. JIMD Rep. 2019; 44: 9-15. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29923087
- Williams M, Valayannopoulos V, Altassan R, Chung WK, Heijboer AC, et al. Clinical, biochemical, and molecular overview of transaldolase deficiency and evaluation of the endocrine function: Update of 34 patients. J Inherit Metab Dis. 2019; 42: 147-158. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30740741
- Loeffen YG, Biebuyck N, Wamelink MM, Jakobs C, Mulder MF, et al. Nephrological abnormalities in patients with transaldolase deficiency. Nephrol Dial Transplant. 2012; 27: 3224-3227. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/22510381
- Vas G, Conkrite K, Amidon W, Qian Y, Bánki K, et al. Study of transaldolase deficiency in urine samples by capillary LC-MS/MS. J Mass Spectrom. 2006; 41: 463-469. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16470722
- Wamelink MM, Struys EA, Jakobs C. The biochemistry, metabolism and inherited defects of the pentose phosphate pathway: a review. J Inherit Metab Dis. 2008; 31: 703-717. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/18987987