
More Information
Submitted: 09 November 2020 | Approved: 18 November 2020 | Published: 19 November 2020
How to cite this article: Yuan K, Wang C. Premature ovarian insufficiency in children: Etiology, clinical management and treatment. J Adv Pediatr Child Health. 2020; 3: 047-055.
DOI: 10.29328/journal.japch.1001017
ORCiD iD: orcid.org/0000-0002-4273-1341
Copyright License: © 2020 Yuan K, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Children; Premature ovarian insufficiency; Etiology; Hormone replacement therapy; Clinical management

Premature ovarian insufficiency in children: Etiology, clinical management and treatment
Ke Yuan and Chunlin Wang*
Department of Pediatrics, The First Affiliated Hospital of Zhejiang University School of Medicine, Hangzhou, Zhejiang Province China
*Address for Correspondence: Chunlin Wang, Department of Pediatrics, The First Affiliated Hospital of Zhejiang University School of Medicine, No: #79 Qingchun Road, Hangzhou, Zhejiang Province, 310003, China, Tel: +86-571-87235128; Email: [email protected]; [email protected]
Premature ovarian insufficiency (POI) is a rare disease, especially in children and adolescents. It was previously called premature ovarian failure (POF). It can be manifested as delayed puberty, primary or secondary amenorrhea that occurred before the age of 40 years with no less than two abnormal serum sex hormones (low estrogen and high gonadotropin). It is reported that the incidence rate is 1% at the age of 40 years and 0.01% at the age of 20 years. Although the disease usually occurs in middle-aged and elderly women, clinical practice in recent years has shown that it has also been found in adolescents and even children. It is generally believed that the etiology of POI includes genetic factors, immune factors, and iatrogenic factors. So far, several genetic mutations that may cause POI have been found clinically, but the etiology of 90% of POI is still unknown. In recent years, the incidence of POI in children and adolescents has increased, and there are more urgent requirements for its early diagnosis, treatment, and clinical management. Based on this, this article will mainly review the research progress of the etiology, treatment, and clinical management of POI in children and adolescents.
Premature ovarian insufficiency (POI) used to be called primary ovarian insufficiency or premature ovarian failure [1]. It refers to a clinical syndrome caused by a significant decline or loss of ovarian function before the age of 40 years [2,3]. The current diagnostic criteria believe that primary or secondary amenorrhea with no less than two abnormal serum sex hormones (low estrogen and high gonadotropin) that occurred before the age of 40 years can be diagnosed [2]. The prevalence of POI in the general population is roughly 25% – 5%, and some reports indicate that 0.01% of 20-year-old young women may have POI [4]. POI has a serious impact on women’s health in all aspects, such as irregular menstruations, anxiety disorders, memory deficits, hot flashes, and night sweats, etc..[5]. POI seriously affects women’s health in all aspects, especially adolescents and children. Losing normal ovarian function before puberty begins will affect her normal life and social intercourse as a woman. Early diagnosis, intervention, and treatment can effectively protect the residual ovarian function, which is of great significance to patients’ personal life, family harmony, and social stability. Therefore, it is of great significance to diagnose and start treatment in early childhood and adolescence. So far, although the medical community around the world has made a lot of progress in the etiology of POI, there is still a large part of the etiology of POI is unknown [2,3]. Therefore, how to effectively diagnose and treat POI in early childhood is particularly important and urgent. This article will mainly review the etiology, treatment, and clinical management of POI in children, to provide a reference for the clinic.
Progress in the etiology of children with POI
The common etiology of POI includes genetic factors, iatrogenic factors, immune factors and environmental factors, but the etiology of more than half of POI patients is still unknown, which is called idiopathic POI [2] (Figure 1).
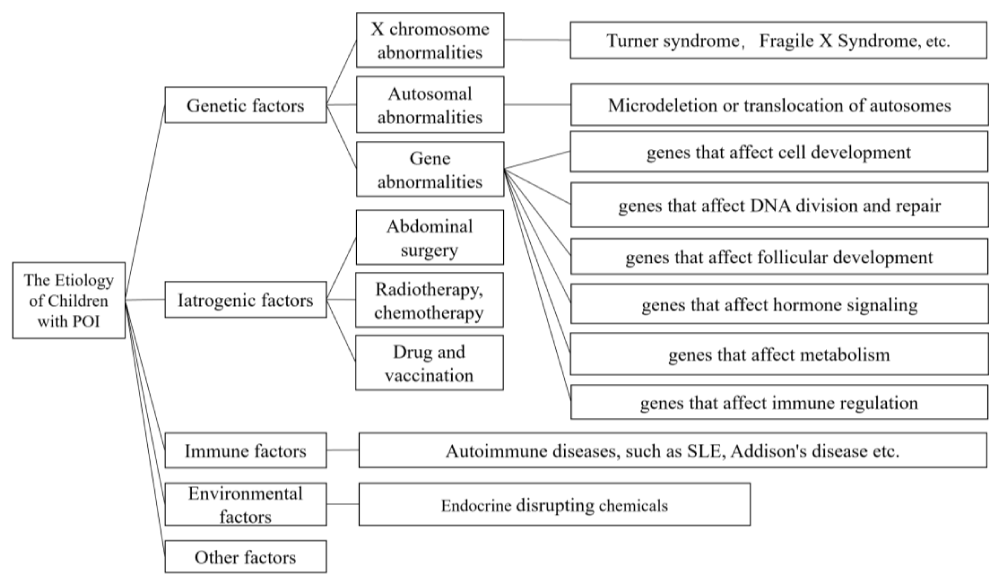
Figure 1: The common etiology of POI includes genetic factors, iatrogenic factors, immune factors and environmental factors, but the etiology of more than half of POI patients is still unknown, which is called idiopathic POI.
Genetic factors
X chromosome abnormalities: Sex chromosome abnormalities, especially X chromosome related abnormalities, are the most common etiology of POI. It accounts for about 10% of all POI patients [2]. These abnormalities usually manifest as partial deletion, complete deletion, translocation or duplication of the X chromosome [6]. The most common etiology of POI in children include Turner syndrome and Fragile X syndrome.
Children with Turner syndrome have only one X chromosome (45, X), and the incidence is about 1/2500 [7]. Most children with this karyotype end in spontaneous abortion during the fetal period, only a few lucky ones can be born [8]. The phenotypes of girls with Turner syndrome usually include height growth restriction, abnormalities in the cardiovascular, lymphatic and renal systems, and other physical characteristics such as webbed necks, thoracic duct lymphatic obstruction, and rapid oocyte apoptosis. Therefore, two X chromosomes are considered necessary for the normal function of the ovaries [9,10]. The mechanism of the disease may be that the dose of a specific X gene product is required for correct ovarian maintenance, or it may be that the oocytes cannot degrade due to the lack of X chromosome homologous partners and normal meiosis [11]. Individuals with trisomy X (47, XXX) usually do not experience reduced fertility, but many case studies report that POI is associated with this karyotype [12,13]. The prevalence of trisomy X in the POI population is estimated to be between 1% to 4% [14].
In addition to Turner syndrome, POI can also be associated with partial chromosomal abnormalities, such as terminal deletions with breakpoints in the proximal Xp and/or proximal Xq regions, such as Xq13 or Xp11 [11]. The key areas for normal ovarian development, Xq13-27 and Xp13-11, have been proposed [15]. These areas may be destroyed by deletions or translocations, both of which may lead to POI. Pathogenic genes in these regions include POF1B and FMR1. In many cases, POI may cause oocyte apoptosis due to unpaired chromosomes during meiosis, but genes in these key regions may also be involved in the pathogenesis of POI. Mutations in the Fragile X Syndrome-related gene FMR1 at Xq27.3 are the second most common congenital etiology of POI [16,17]. Fragile X syndrome is the most common genetic dementia and the most common genetic cause of autism and mental retardation [16]. It is caused by the loss of function of the FMR1 gene. Its manifestations may be mild learning and mental disorders, autism, or even severe mental retardation. This mutation can account for 6% to 13% of POI patients [16]. The 5’UTR region of the gene contains an expandable CGG continuous repeat region. In normal people, this continuous repeat is generally repeated about 45 times, and the amount of repeats in individuals with complete mutations may exceed about 200 times. CGG mutations with a repetitive amount in the range of 55-199 are called pre-mutations and have been confirmed in previous experiments to be associated with the risk of POI in women [18].
Autosomal abnormalities: In recent years, some POI caused by autosomal abnormalities have been reported frequently, but so far there is no clear and functional verification of related genes. It is generally believed that the microdeletion, translocation, etc. of autosomes such as 13, 14, 15, 18, and 21 may cause POI. These regions are rich in genes, such as BAX, BCL2, CDKN1B, CYP19A1, ESR1, FOXL2, CASP2, and CASP3, and other related genes [19-23]. It can participate in the control of follicle apoptosis and regulation, which will be reviewed in detail in the next section.
Gene abnormalities: Gene abnormalities can affect ovarian function in different ways. Common ones include genes that affect development, genes that affect DNA division and repair, genes that affect follicular development and hormone signaling, genes that affect metabolism, and genes that affect immune regulation [24].
The function of the ovary depends on a reasonable ovarian structure to effectively accommodate, support, and protect the developing and well-developed germ cells. It is currently known that some genes influence the production and prognosis of POI by participating in the occurrence of gonads. These include NR5A1, FOXL2, BMPR1B, and other related genes [25]. The NR5A1 gene can encode steroid-producing factor-1, which can regulate adrenal gland and reproductive system development. Mutations in the NR5A1 gene can cause a series of gonadal dysfunction, including POI and 46, XX gonadal hypoplasia [26]. The FOXL2 gene is another gene required for gonad genesis. It is expressed in the development of ovarian follicles and eyelids and is characterized by POI patients with or without related eyelid deformities. The FOXL2 gene is not only particularly important in the development of ovarian follicles, but also necessary for maintaining a normal physiological state after ovarian development is complete [27]. Through extensive studies on mice, goats, and humans, the importance of FOXL2 gene expression throughout development and adulthood has been well confirmed, and the specific cells and periods of its influence can be located. The ovarian phenotypes of women carrying the FOXL2 gene are diverse. Some clinical studies have reported the existence of this difference. Compared with normal women, the possibility of ovarian cysts is more likely to occur in groups with FOXL2 gene mutations [28-30]. In conclusion, patients with mutations in the FOXL2 gene may have different ovarian phenotypes or follicular defects. The BMPR1B gene is the receptor of GDF, which is of great significance for the development of gonads and bones. At present, clinical studies have found that BMPR1B gene mutations lead to ovarian loss or dysplasia with local achondroplasia [31]. Some other genes can affect the development of the follicle and lead to the production of POI by affecting the development of the internal structure of the ovary, the outer barrier of the follicle, and the initial and mature stages of the follicle, waiting for more clinical findings to be confirmed.
If the ovaries develop normally and maintain normal physiological functions, the dysfunction of oocytes is an important etiology of POI. Normal meiosis of follicular cells is very important for normal ovarian function. Different genes can affect the process of meiosis by influencing the changes in different periods of meiosis and then affect the normal ovarian function, such as the correct arrangement of chromosomes, the normal function of spindle fiber tissue, and the process of chromosome separation to both ends. Both may lead to abnormal meiosis. For example, cohesin protein is an important protein needed to affect sister chromatid aggregation during meiosis. Mutations in the STAG3 gene can cause cohesin protein abnormalities. This disease was recently confirmed in a Lebanese family. Two girls in this family had primary amenorrhea due to mutations in the STAG3 gene and had no secondary sexual characteristics at all [32]. The same is true of the loss of integrity of the cohesive complex caused by mutations in the SGO2 gene [33]. The POF1B gene edits a myosin-like protein that interacts with non-muscle actin filaments to affect the division process of meiotic cells. The POF1B gene is located in the long arm of the X chromosome, which is a region known to be critical to ovarian function. At present, POI case reports caused by mutations in this gene have also been discovered [34].
During meiosis, homologous chromosomal recombination occurs before the chromosomes produce synaptic complexes. The destruction of the synaptic complex in mice can lead to infertility [35]. This principle also applies to humans. Recent experiments have confirmed that mutations in the SYCE1 subunit of the complex can lead to the production of human POI. Recent studies have also found that mutations in genes required for homologous recombination, such as HFM1 and PSMC3IP genes, may lead to the production of human POI [36]. Besides, it was reported that XRCC gene mutation can also lead to POI through the effect of meiotic homologous recombination, but the reporter did not conduct further research [37]. The cell division errors caused by the above-mentioned gene disruption at different stages of meiosis may lead to the production of abnormal oocytes, and then these abnormal oocytes often undergo programmed death during development. Therefore, there is an effective DNA damage repair mechanism in the process of maintaining normal human meiosis. The most common DNA damage repair genes associated with POI are ATM genes. ATM is one of the kinases for cell cycle checking, and cells must respond to DNA damage. ATM gene mutations can cause ataxia, including cerebellar degeneration, oculomotor nerve dysfunction, immune deficiency, increased sensitivity, chromosomal instability, gonadal abnormalities, and decreased germ cells [38]. Recently, many gene mutations related to DNA damage repair have been discovered, such as MCM8 and MCM9, two genes encoding chromosome maintenance proteins, which are necessary for double-stranded DNA break repair. MCM8 gene mutation can cause POI with hypothyroidism, and MCM9 gene mutation can cause POI with short stature [39,40].
The CSB-PGBD3 fusion gene encodes a transcription-coupled DNA repair protein, and its mutation can also cause POI [41]. Also, in a normal ovulation cycle, only one dominant follicle will be discharged normally every month. Therefore, genes involved in apoptosis may also cause the production of POI. Some mouse experimental models have also confirmed this conjecture. For example, the NANOS3 gene plays a direct role in inhibiting the apoptosis of migrating primordial germ cells, so its gene mutation may directly lead to the production of POI. Previous studies have proved that progesterone has a clear anti-apoptotic effect in ovarian cells, so mutations in the PGRMC1 gene that affect the composition of progesterone receptor ink can also lead to the production of POI patients. Genes involved in protein translation are also very important for the growth and development of oocytes. Mutations in key components of the translation process lead to apoptosis and increased atresia, which is also an important reason for POI. For example, the editing of translation initiation factors elF2B2, elF2B4, and elF2B5 gene mutations may lead to POI in patients with leukodystrophy. In this regard, there are other mutations in the RCBTB1 gene, elF4EN1F1 gene, etc., which may cause POI [42].
The production, development, and maturation of oocytes are also inseparable from the participation of various factors. These include growth factors such as BMP, GDF, and neurotrophic factors such as NGF. BPM15 is an oocyte-specific growth factor, which is located at the Xp locus closely related to ovarian reserve. The biological tasks of BMP15 include promoting the growth and maturation of follicles, regulating the sensitivity of follicular granulosa cells to FSH and the production of regular ovulation rhythms, and preventing granulosa cell apoptosis [43]. Current research has determined that mutations in the BMP15 gene can cause POI. Other members of the BMP family, such as BMP4 or BMP7, also play an important role in the hormonal control of follicle formation, which may also lead to the production of POI, but there is currently no real evidence [44]. Noggin protein is a protein that combines BMP4 and BMP7. It is also important for the development of ovaries and bones. The mutation of the NOG gene is positively related to the proximal end of POI. Another gene involved in the formation of follicles is GDF9. Experiments in mice confirmed that normal follicles in mice with damaged GDF9 gene remained in the primary stage. Some studies on the GDF9 gene also confirmed that this gene may be related to the production of POI [45].
NOBOX gene is an oocyte-specific homologous gene, which can be used as a transcriptional regulator of many different types of ovarian genes, including the GDF9 and BMP15. It has been identified as a marker of oocytes. It is expressed in germ cell cysts and primordial and growing oocyte banks. In mice that knocked out the NOBOX gene, this factor did cause rapid apoptosis of oocytes after birth and blocked the process of primordial follicles to mature follicles. Many cases of POI caused by NOBOX gene defects have been reported, and they all appear to be heterozygous [46]. Compared with animal models of knockout genes, the clinical symptoms of patients are often not serious, which may be related to gene heterozygosity. Functional studies have shown that due to the defect of the NOBOX gene, it can inhibit the transcriptional activity of GDF9 by more than 50%. The prevalence of NOBOX in the POI population exceeds 5%, which may be one of the most common POI genetic defects. This shows its key position in the development of human follicles.
Since the growth and development of follicles are heavily dependent on hormones, many genes involved in POI production are involved in mediating hormone signal transduction. The most classic of these is the key hormone that promotes the growth and development of follicles-FSH. FSH contains two different subunits, one is the α subunit shared with human chorionic gonadotropin, and the other is the FSH-specific β subunit [47]. Mutations in the FSH receptor FSHR are associated with a variety of clinical POI phenotypes, depending on the residual function of the mutant receptor. FSHR is a gene that has a certain connection between a few specific mutations and a specific phenotype. For example, the homozygous p.A189V mutation may cause female primary amenorrhea and follicular dysplasia or damage. When women with this mutation are treated with high-dose FSH, there is rarely a functional response. Individuals with compound heterozygous FSHR mutations often cause an only partial loss of function. These mutations are related to the new phenotype. For example, these women can have normal puberty, normal-sized ovaries, and have seemingly normal ovaries. Follicles. However, the FSH levels of these women are quite high, and these seemingly normal follicles often die in the later stages of development [47]. Therefore, understanding the residual function of FSHR is likely to help us predict the phenotype of children or adolescent women in adulthood, and formulate a reasonable treatment plan, which requires further research. Estrogen has important significance in female reproductive health, including the development of secondary sexual characteristics and the regulation of the ovulation cycle. In women, the synthesis of estrogen begins when cholesterol enters the mitochondria of follicular cells and is converted into pregnenolone by CYP11 enzyme, then pregnenolone is converted into androgen by CYP17 enzyme, and finally by granule cells by CYP19 enzyme convert these androgens into estrogen. All genes involved in this process may lead to the production of POI. And a large part of it has been confirmed in clinical practice. The STAR protein is responsible for transferring cholesterol to the mitochondria to produce steroids, while the CYP17A1 and CYP19A1 genes encode enzymes for the aromatization of pregnenolone and estrogen, respectively. Mutations in these genes may cause POI [48].
Many reported genetic defects in POI patients are involved in the establishment of mitochondrial function. Many studies have shown that the absence of POI patients has significantly elevated oxidative stress markers. Human oocytes contain about 100,000 copies of mtDNA, which is significantly more than normal somatic cells. Oocytes may require a high copy number of mtDNA to support the fertilization process and embryonic development before implantation into the uterine wall [49]. Women with POI have significantly lower mtDNA levels, and this decrease can be detected in the blood. The PLOG gene is a gene encoding mtDNA polymerase, which is necessary for the effective and accurate replication of mtDNA. Mutations in this gene may cause consumption or deletion of mtDNA, which directly leads to POI with blindness, myopathy, or Parkinson’s symptoms. Similarly, patients with mutations in the C100rf2 gene can also cause the loss of mtDNA and lead to POI with deafness, which is called Perrault syndrome. Other similar genes include LARS2, HARS2, and AARS2 genes, etc., which achieve the purpose of disease generation by disrupting the homeostasis of mitochondria [50-52].
There are many genes involved in the pathogenesis of POI, but there are few clear explanations of the mechanism or mode of action. For example, many autoimmune diseases, such as systemic lupus erythematosus, Hashimoto’s thyroiditis often occur simultaneously with POI. This may be related to the effect of autoantibodies produced by autoimmune diseases on ovarian and follicular membrane cells [38]. There is little research on the molecular mechanism of autoimmune-mediated ovarian diseases. There is also one of the most common genetic factors for POI, Fragile X Syndrome, which we have mentioned in the previous section related to the X chromosome. This is one of the most common causes of POI in children. But again, the mechanism research is still unclear.
Iatrogenic factors
Abdominal surgery, radiotherapy in cancer treatment, chemotherapy, drug application, and vaccination may all cause POI in children. Iatrogenic injuries to the pelvis, ovaries, uterus, and surrounding soft tissues may affect the blood supply of the ovaries or other organs to cause POI.
Different from the large proportion of adult surgery, radiotherapy, and chemotherapy in the iatrogenic factors of POI, children should pay more attention to the possibility of POI caused by the application of drugs and vaccination. A recent report mentioned that girls after HPV vaccination may cause POI [53]. This is different from our previous understanding of HPV vaccines. Many vaccines have not passed the research on the impact on the gonads before they are on the market. Especially for vaccines for the diagnosis of children and adolescents, their safety should be further improved. In terms of drugs, the worthiest of vigilance is the use of immunosuppressants or biological agents in the treatment of immune-related diseases. Some studies have found that in the treatment of SLE, the massive use of cyclophosphamide may lead to the production of POI. Cyclophosphamide is a common drug for the treatment of SLE, and the potential gonadal risk of more biological agents is not yet clearly recognized [54]. In the process of treating autoimmune-related diseases, how to control the dosage of immunosuppressants, and avoid the side effects of gonadal toxicity requires more in-depth research.
With the high incidence of certain cancers in recent years, many children have begun to receive radiotherapy and chemotherapy for their illnesses, which is of great significance to their lives. However, the younger the treatment begins, the greater the damage to the ovaries by these radiations or chemicals. Oocytes are cells that are very sensitive to radiation. Repeated radiotherapy can cause premature follicle apoptosis [1]. Therefore, in the process of treatment, the protection of the abdomen is particularly important, to reduce the damage to the gonads, to achieve the purpose of reducing the production of POI.
Immune factors
Many autoimmune diseases, such as SLE, Hashimoto’s thyroiditis, Addison’s disease, myasthenia gravis, ankylosing spondylitis, autoimmune polyglandopathy, etc. often occur simultaneously with POI. This may be related to the effect of autoantibodies produced by autoimmune diseases on ovarian and follicular membrane cells. Autoimmune oophoritis is related to mononuclear inflammatory cell infiltration of follicular membrane cells and autoantibodies against ovaries, steroid production, or adrenal cortex tissue [38]. Autoimmune POI is almost always associated with Addison’s disease, and affected women can usually be tested positive for autoantibodies against steroid-producing enzymes, such as 21-hydroxylase and 17-hydroxylase [38]. Little is known about the molecular mechanisms leading to autoimmune-mediated ovarian autoimmunity. Autoimmune diseases usually have a genetic component. The research in this area needs to be further deepened.
Environmental factors
Environmental factors that can affect the production of POI has been confirmed in many experiments or clinical observations. These influencing factors are called endocrine-disrupting chemicals (EDCs)[55]. EDCs can induce follicular atresia by causing oxidative stress and apoptosis in the human body, such as BPA, phthalates, PAH, cigarette smoke, pesticides, dioxins, genistein, PCB. Second, exposure to certain substances may lead to a decrease in the number of primordial follicles, such as PAHs, DEHP, cigarette smoke, genistein, and BPA[55]. For example, exposure to DEHP before puberty significantly reduced the percentage of antral follicles and increased the number of messenger RNA of Pro-apoptotic genes. Oxidative stress and ovarian somatic cell apoptosis induced by DEHP. The toxic effect of phthalates on the ovary rests on folliculogenesis and steroidogenesis disorders with, as a consequence, an alteration in reproductive functions, infertility and POI. BPA is an endocrine disruptor acting notably like an estrogen mimicker on the estrogen receptor α. Exposure to BPA leads to a decrease in the number of primordial follicles. Besides, some harmful substances widely distributed in the environment such as heavy metals such as mercury, arsenic, and cadmium can also cause POI by damaging the ovarian tissue. These environmental pollutants pose a serious threat to human and animal reproduction and have harmful effects that interfere with endocrine and reproductive functions. This is a key public health issue that requires protection, prevention, and remedial measures to combat the harm caused by these environmental pollutants.
Other factors
Other factors that may cause POI to include infection, personal life, and idiopathic factors. The specific mechanism needs to be further studied.
In addition to causing infertility, POI in children is also associated with a variety of health risks, including decreased bone density and increased fracture risk, and early progression of cardiovascular disease, psychological effects, including depression, anxiety and decreased perceived psychosocial support, and potential recognition. Knowing the early decline and so on, so early treatment and long-term management are particularly important [56].
Hormone replacement therapy (HRT)
HRT is the main method of treatment for children with POI. Currently, the commonly used HRT methods include transdermal, vaginal, and oral administration. Evidence shows that transdermal or transvaginal estradiol therapy is the first-line treatment option for children with POI. These young women need long-term ovarian sex steroid replacement, which generally requires this therapy for decades, until the age of normal menopause [57].
Ideally, hormone replacement will mimic normal ovarian function. These formulations mimic the daily rate of ovarian production of estradiol and bring the average serum estradiol level to the level of normal women, which is the average level of women with normal ovarian function throughout the menstrual cycle [58]. The equivalent dose of oral estradiol is also an effective alternative. However, transdermal and transvaginal routes deliver the hormone directly into the circulation, which avoids complications related to the first-pass effect of the liver when estrogen is taken orally. Compared with transdermal estrogen use, oral estrogen can increase the risk of venous thromboembolism. Unlike oral estrogen, transdermal HRT does not adversely affect cardiovascular disease or thromboembolism risk markers, and it also avoids the psychological barriers of vaginal administration to children. In women with underlying obesity or coagulopathy, the risk of venous thromboembolism associated with oral estrogen is further increased, which is 5-8 times the risk seen by non-users or users of transdermal estrogen preparations [59]. And small doses of estrogen will not hurt children’s height growth. For specific treatment programs for children, low-dose 17-beta estradiol is generally chosen to induce puberty at about 12 years old, and then the dose is gradually increased according to the physiological changes of girls in the next 2 to 3 years. After estrogen is used for at least 2 years or after bleeding occurs, progesterone is periodically added for endometrial protection to stimulate menstruation. For children who are diagnosed with POI later or who no longer need to consider the effects of height growth and development, cyclic estrogen and progesterone can be directly given co-treatment [2].
Management of cardiovascular health
The shortened life expectancy of untreated POI patients is mainly due to the occurrence of cardiovascular disease [60]. Compared with the age-matched control group, women with POI have reduced vascular endothelial function, which is an early sign of atherosclerosis. Using HT for 6 months in this group of women significantly improved endothelial function. Estrogen is beneficial to cholesterol metabolism, can reduce atherosclerotic plaque formation, and prevent coronary artery contraction through catecholamine regulation. Since estrogen deficiency is the culprit, it is recommended to start using HRT as a cardioprotective strategy early. Although the data from randomized controlled trials on the cardiovascular benefits of HRT are still unclear, experts in the field agree that in otherwise healthy women with POI, the initiation and continuation of HRT is of great significance to the patients themselves, until the average age of menopause is achieved. The benefits far outweigh any so-called risks[61]. Although longitudinal outcome data are lacking for POI in children and adolescents, the cardioprotective effect of HRT can be inferred from data from postmenopausal women. Besides, for POI women, avoiding related risk factors (such as healthy diet, proper exercise, and refusal to smoke) to ensure cardiovascular health is crucial.
Management of bone health
Estrogen deficiency in POI is an important factor leading to fractures, and this risk can be alleviated by using HRT. Compared with menstruating women of the same age, POI patients not only have a significantly lower bone density, but 21% of POI patients have a BMD z-score of less than -2.0, putting them at significant risk of fracture, and it will gradually increase with age aggravate [62]. The appearance of low bone mass should be considered as an indication for priority initiation of HRT in any woman diagnosed with POI, and in these patients, continuous monitoring of bone mass (within 2 to 5 years of initiation of HRT) is required. HRT may have a greater impact on children, especially if they have not yet reached their peak bone mass. Lifestyle changes, such as weight-bearing exercise, optimization of dietary calcium intake and vitamin D status, and avoiding smoking can all help maintain bone health.
Management of emotion
The diagnosis of POI is usually unexpected, especially for children. The life process of a normal woman tends to end before it begins, which is unacceptable to most children. Impaired fertility, fear of premature aging, and being different from people of the same age may all be emotionally unable to cope. Compared with normal peers, children with POI have weaker self-esteem, increased social anxiety and shyness, and significantly worsened depressive symptoms [63]. For any child with POI, early psychological counseling should be promoted and made accessible. Frequent visits with the female multidisciplinary team should be arranged to continuously observe her coping strategies and address unmet needs.
Assisted reproductive technology (ART)
Some children with POI have the possibility of natural ovulation, which allows them to become mothers in the future. For children, the early diagnosis of POI and the cryopreservation of oocytes or ovarian tissue may preserve possible reproductive capacity. There have been fetuses of this technology, but technical barriers and huge capital investment limit the possibility of clinical application in this area.
POI is a disease that has a serious impact on women, especially children. Delay in diagnosis and treatment may greatly affect the health and well-being of children in the future. The etiology of POI is complex, and more than half of the causes of POI are currently unclear. Clinically, multi-factor investigation of POI diseases should be carried out as early as possible to achieve early detection and early treatment. Primary POI is mainly manifested in children with POI, and the main cause is genetic factors. The current diagnostic criteria for POI still lag the mechanism research. Pediatric endocrinologists or pediatric and female physicians should pay attention to the use of a variety of diagnostic methods during the examination of children with POI and diagnose as soon as possible to achieve the best treatment effect.
Funding
Fully funded by the key Research and Development Program of Zhejiang Province with grant number: 2020C03121.
- Goswami D, Conway GS. Premature ovarian failure. Horm Res. 2007: 68: 196-202. PubMed: https://pubmed.ncbi.nlm.nih.gov/17495481/
- Webber L, Davies M, Anderson R, Bartlett J, Braat D, et al. ESHRE Guideline: management of women with premature ovarian insufficiency. Hum Reprod. 2016; 31: 926-397. PubMed: https://pubmed.ncbi.nlm.nih.gov/27008889/
- Vujovic S, Ivović M, Tancić-Gajić M, Marina L, Barać M, et al. Premature ovarian failure. Srp Arh Celok Lek. 2012; 140: 806-811. PubMed: https://pubmed.ncbi.nlm.nih.gov/23350261/
- Tao XY, Zuo AZ, Wang JQ, Tao FB. Effect of primary ovarian insufficiency and early natural menopause on mortality: a meta-analysis. Climacteric. 2016; 19: 27-36. PubMed: https://pubmed.ncbi.nlm.nih.gov/26576012/
- Tsiligiannis S, Panay N, Stevenson JC. Premature Ovarian Insufficiency and Long-Term Health Consequences. Curr Vasc Pharmacol. 2019; 17: 604-609. PubMed: https://pubmed.ncbi.nlm.nih.gov/30819073/
- Laissue P. Aetiological coding sequence variants in non-syndromic premature ovarian failure: From genetic linkage analysis to next generation sequencing. Mol Cell Endocrinol. 2015; 411: 243-257. PubMed: https://pubmed.ncbi.nlm.nih.gov/25960166/
- Ferreira SI, Matoso E, Pinto M, Almeida J, Liehr T, et al. X-chromosome terminal deletion in a female with premature ovarian failure: Haploinsufficiency of X-linked genes as a possible explanation. Mol Cytogenet. 2010; 3: 14. PubMed: https://pubmed.ncbi.nlm.nih.gov/20646274/
- Hook EB, Warburton D. Turner syndrome revisited: review of new data supports the hypothesis that all viable 45,X cases are cryptic mosaics with a rescue cell line, implying an origin by mitotic loss. Hum Genet. 2014; 133: 417-424. PubMed: https://pubmed.ncbi.nlm.nih.gov/24477775/
- Modi D, Sane S, Bhartiya D. Over-expression of Mullerian inhibiting substance mRNA in the Turner syndrome ovary. Sex Dev. 2009; 3: 245-252. PubMed: https://pubmed.ncbi.nlm.nih.gov/19940443/
- Karnis MF. Catastrophic consequences of assisted reproduction: the case of Turner syndrome. Semin Reprod Med. 2012; 30: 116-122. PubMed: https://pubmed.ncbi.nlm.nih.gov/22549711/
- Simpson JL, Rajkovic A. Ovarian differentiation and gonadal failure. Am J Med Genet. 1999; 89: 186-200. PubMed: https://pubmed.ncbi.nlm.nih.gov/10727994/
- Kara C, Üstyol A, Yılmaz A, Altundağ E, Oğur G. Premature ovarian failure due to tetrasomy X in an adolescent girl. Eur J Pediatr. 2014; 173: 1627-1630.
- Sugawara, N, Maeda M, Manome T, Nagai R, Araki Y. Patients with 47, XXX karyotype who experienced premature ovarian failure (POF): two case reports. Reprod Med Biol. 2013; 12: 193-195. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5906948/
- Wood A, Kleis L, Toriello H, Cemeroglu AP. Mosaic pentasomy X/tetrasomy X syndrome and premature ovarian failure. Indian Pediatr. 2011; 48: 402-404. PubMed: https://pubmed.ncbi.nlm.nih.gov/21654007/
- Therman E, Laxova R, Susman B. The critical region on the human Xq. Hum Genet. 1990; 85: 455-461. PubMed: https://pubmed.ncbi.nlm.nih.gov/2227929/
- Conway GS. Premature ovarian failure and FMR1 gene mutations: an update. Ann Endocrinol (Paris). 2010; 71: 215-217. PubMed: https://pubmed.ncbi.nlm.nih.gov/20398889/
- Bouali, N, Hmida D, Mougou S, Bouligand J, Lakhal B, et al. Analysis of FMR1 gene premutation and X chromosome cytogenetic abnormalities in 100 Tunisian patients presenting premature ovarian failure. Ann Endocrinol (Paris). 2015; 76: 671-678. PubMed: https://pubmed.ncbi.nlm.nih.gov/26593861/
- Pastore LM, Johnson J. The FMR1 gene, infertility, and reproductive decision-making: a review. Front Genet. 2014; 5: 195. PubMed: https://pubmed.ncbi.nlm.nih.gov/25071825/
- Yang XW, He W, Gong F, Li W, Li X, et al. Novel FOXL2 mutations cause blepharophimosis-ptosis-epicanthus inversus syndrome with premature ovarian insufficiency. Mol Genet Genomic Med. 2018; 6: 261-267. PubMed: https://pubmed.ncbi.nlm.nih.gov/29378385/
- Chen B, Li L, Wang J, Li T, Pan H, et al. Consanguineous familial study revealed biallelic FIGLA mutation associated with premature ovarian insufficiency. J Ovarian Res. 2018; 11: 48. PubMed: https://pubmed.ncbi.nlm.nih.gov/29914564/
- Franic-Ivanisevic M, Franić D, Ivović M, Tančić-Gajić M, Marina L, et al. Genetic Etiology of Primary Premature Ovarian Insufficiency. Acta Clin Croat. 2016; 55: 629-635. PubMed: https://pubmed.ncbi.nlm.nih.gov/29117655/
- Jaillard S, Tucker SJ, Akloul L, Beaumont M, Domin M, et al. 22q11.2 rearrangements found in women with low ovarian reserve and premature ovarian insufficiency. J Hum Genet. 2018; 63: 691-698. PubMed: https://pubmed.ncbi.nlm.nih.gov/29540854/
- Wang Q, Li D, Cai B, Chen Q, Li C, et al. Whole-exome sequencing reveals SALL4 variants in premature ovarian insufficiency: an update on genotype-phenotype correlations. Hum Genet. 2019; 138: 83-92. PubMed: https://pubmed.ncbi.nlm.nih.gov/30603774/
- Tucker EJ, Grover SR, Bachelot A, Touraine P, Sinclair AH. Premature Ovarian Insufficiency: New Perspectives on Genetic Cause and Phenotypic Spectrum. Endocr Rev. 2016; 37: 609-635. PubMed: https://pubmed.ncbi.nlm.nih.gov/27690531/
- Lin L, Achermann JC. Steroidogenic factor-1 (SF-1, Ad4BP, NR5A1) and disorders of testis development. Sex Dev. 2008; 2: 200-209. PubMed: https://pubmed.ncbi.nlm.nih.gov/18987494/
- Brauner R, Pierrepont S, Bignon-Topalovic J, McElreavey K, Bashamboo A. Etiology of primary ovarian insufficiency in a series young girls presenting at a pediatric endocrinology center. Eur J Pediatr. 2015; 174: 767-773. PubMed: https://pubmed.ncbi.nlm.nih.gov/25425520/
- Irusta G, Pazos MC, Abramovich D, De Zúñiga I, Parborell F, et al. Effects of an inhibitor of the gamma-secretase complex on proliferation and apoptotic parameters in a FOXL2-mutated granulosa tumor cell line (KGN). Biol Reprod. 2013; 89: 9. PubMed: https://pubmed.ncbi.nlm.nih.gov/23699387/
- Li Y, Schang G, Wang Y, Zhou X, Levasseur A, et al. Conditional Deletion of FOXL2 and SMAD4 in Gonadotropes of Adult Mice Causes Isolated FSH Deficiency. Endocrinology. 2018; 159: 2641-2655. PubMed: https://pubmed.ncbi.nlm.nih.gov/29800110/
- Martinez-Juarez A, López-Luna MA, Porras-Gómez TJ, Moreno-Mendoza N. Expression of the Sox9, Foxl2, Vasa, and TRPV4 genes in the ovaries and testes of the Morelet's crocodile, Crocodylus moreletii. J Exp Zool B Mol Dev Evol. 2018; 330: 148-164. PubMed: https://pubmed.ncbi.nlm.nih.gov/29602213/
- Tang B, Zhang Y, Zhang W, Zhu Y, Yuan S. Deletion of FOXL2 by CRISPR promotes cell cycle G0/G1 restriction in KGN cells. Int J Mol Med. 2019; 43: 567-574. PubMed: https://pubmed.ncbi.nlm.nih.gov/30365048/
- Stange K, Désir J, Kakar N, Mueller TD, Budde BS, et al. A hypomorphic BMPR1B mutation causes du Pan acromesomelic dysplasia. Orphanet J Rare Dis. 2015; 10: 84. PubMed: https://pubmed.ncbi.nlm.nih.gov/26105076/
- Caburet S, Arboleda VA, Llano E, Overbeek PA, Barbero JS, et al. Mutant cohesin in premature ovarian failure. N Engl J Med. 2014; 370: 943-949. PubMed: https://pubmed.ncbi.nlm.nih.gov/24597867/
- Faridi R, Rehman AU, Morell RJ, Friedman PL, Demain L, et al. Mutations of SGO2 and CLDN14 collectively cause coincidental Perrault syndrome. Clin Genet. 2017; 91: 328-332. PubMed: https://pubmed.ncbi.nlm.nih.gov/27629923/
- Lacombe A, Lee H, Zahed L, Choucair M, Muller J, et al. Disruption of POF1B binding to nonmuscle actin filaments is associated with premature ovarian failure. Am J Hum Genet. 2006; 79: 113-119. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1474115/
- de Vries FA, de Boer E, van den Bosch M, Baarends WM, Ooms M, et al. Mouse Sycp1 functions in synaptonemal complex assembly, meiotic recombination, and XY body formation. Genes Dev. 2005; 19: 1376-1389. PubMed: https://pubmed.ncbi.nlm.nih.gov/15937223/
- Al-Agha AE, Ahmed IA, Nuebel E, Moriwaki M, Moore B, et al. Primary Ovarian Insufficiency and Azoospermia in Carriers of a Homozygous PSMC3IP Stop Gain Mutation. J Clin Endocrinol Metab. 2018; 103: 555-563. PubMed: https://pubmed.ncbi.nlm.nih.gov/29240891/
- Zhang YX, Li H, He W, Tu C, Du J, et al. XRCC2 mutation causes premature ovarian insufficiency as well as nonobstructive azoospermia in humans. Clin Genet. 2019; 95: 442-443. PubMed: https://pubmed.ncbi.nlm.nih.gov/30489636/
- De Bellis A, Bellastella G, Falorni A, Aitella E, Barrasso M, et al. Natural history of autoimmune primary ovarian insufficiency in patients with Addison's disease: from normal ovarian function to overt ovarian dysfunction. Eur J Endocrinol. 2017; 177: 329-337. PubMed: https://pubmed.ncbi.nlm.nih.gov/28733292/
- AlAsiri S, Basit S, Wood-Trageser MA, Yatsenko SA, Jeffries EP, et al. Exome sequencing reveals MCM8 mutation underlies ovarian failure and chromosomal instability. J Clin Invest. 2015; 125: 258-262. PubMed: https://pubmed.ncbi.nlm.nih.gov/25437880/
- Goldberg Y, Halpern N, Hubert A, Adler SN, Cohen S, et al. Mutated MCM9 is associated with predisposition to hereditary mixed polyposis and colorectal cancer in addition to primary ovarian failure. Cancer Genet. 2015; 208: 621-624. PubMed: https://pubmed.ncbi.nlm.nih.gov/26806154/
- Qin Y, Guo T, Li G, Tang T, Zhao S, et al. CSB-PGBD3 Mutations Cause Premature Ovarian Failure. PLoS Genet. 2015; 11: e1005419. PubMed: https://pubmed.ncbi.nlm.nih.gov/26218421/
- Kasippillai T, MacArthur DG, Kirby A, Thomas B, Lambalk CB, et al. Mutations in eIF4ENIF1 are associated with primary ovarian insufficiency. J Clin Endocrinol Metab. 2013; 98: E1534-1539. PubMed: https://pubmed.ncbi.nlm.nih.gov/23902945/
- Persani L, Rossetti R, Di Pasquale E, Cacciatore C, Fabre S. The fundamental role of bone morphogenetic protein 15 in ovarian function and its involvement in female fertility disorders. Hum Reprod Update. 2014; 20: 869-883. PubMed: https://pubmed.ncbi.nlm.nih.gov/24980253/
- Regan S, Knight PG, Yovich JL, Leung Y, Arfuso F, et al. Involvement of Bone Morphogenetic Proteins (BMP) in the Regulation of Ovarian Function. Vitam Horm. 2018; 107: 227-261. PubMed: https://pubmed.ncbi.nlm.nih.gov/29544632/
- Belli M, Shimasaki S. Molecular Aspects and Clinical Relevance of GDF9 and BMP15 in Ovarian Function. Vitam Horm. 2018; 107: 317-348. PubMed: https://pubmed.ncbi.nlm.nih.gov/29544636/
- Qin Y, Shi Y, Zhao Y, Carson SA, Simpson JL, et al. Mutation analysis of NOBOX homeodomain in Chinese women with premature ovarian failure. Fertil Steril. 2009; 91(4 Suppl): 1507-1509. PubMed: https://pubmed.ncbi.nlm.nih.gov/18930203/
- Kuechler A, Hauffa BP, Köninger A, Kleinau G, Albrecht B, et al. An unbalanced translocation unmasks a recessive mutation in the follicle-stimulating hormone receptor (FSHR) gene and causes FSH resistance. Eur J Hum Genet. 2010; 18: 656-661. PubMed: https://pubmed.ncbi.nlm.nih.gov/20087398/
- Simsek E, Ozdemir I, Lin L, Achermann JC. Isolated 17,20-lyase (desmolase) deficiency in a 46,XX female presenting with delayed puberty. Fertil Steril. 2005; 83: 1548-1551. PubMed: https://pubmed.ncbi.nlm.nih.gov/15866602/
- Pagnamenta AT, Taanman J, Wilson CJ, Anderson NE, Marotta R, et al. Dominant inheritance of premature ovarian failure associated with mutant mitochondrial DNA polymerase gamma. Hum Reprod. 2006; 21: 2467-2473. PubMed: https://pubmed.ncbi.nlm.nih.gov/16595552/
- Hornig-Do HT, Montanari A, Rozanska A, Tuppen HA, Almalki AA, et al. Human mitochondrial leucyl tRNA synthetase can suppress non cognate pathogenic mt-tRNA mutations. EMBO Mol Med. 2014; 6: 183-193. PubMed: https://pubmed.ncbi.nlm.nih.gov/24413189/
- Pierce SB, Gersak K, Michaelson-Cohen R, Walsh T, Lee MK, et al. Mutations in LARS2, encoding mitochondrial leucyl-tRNA synthetase, lead to premature ovarian failure and hearing loss in Perrault syndrome. Am J Hum Genet. 2013; 92: 614-620. PubMed: https://pubmed.ncbi.nlm.nih.gov/23541342/
- Dallabona C, Diodato D, Kevelam SH, Haack TB, Wong LJ, et al. Novel (ovario) leukodystrophy related to AARS2 mutations. Neurology. 2014; 82: 2063-2071. PubMed: https://pubmed.ncbi.nlm.nih.gov/24808023/
- Little DT, Ward HR. Adolescent Premature Ovarian Insufficiency Following Human Papillomavirus Vaccination: A Case Series Seen in General Practice. J Investig Med High Impact Case Rep. 2014; 2: 2324709614556129. PubMed: https://pubmed.ncbi.nlm.nih.gov/26425627/
- Akawatcharangura P, Taechakraichana N, Osiri M. Prevalence of premature ovarian failure in systemic lupus erythematosus patients treated with immunosuppressive agents in Thailand. Lupus. 2016; 25: 436-444. PubMed: https://pubmed.ncbi.nlm.nih.gov/26621134/
- Vabre P, Gatimel N, Moreau J, Gayrard V, Picard-Hagen N, et al. Environmental pollutants, a possible etiology for premature ovarian insufficiency: a narrative review of animal and human data. Environ Health. 2017; 16: 37. PubMed: https://pubmed.ncbi.nlm.nih.gov/28388912/
- Torrealday S, Kodaman, Pal L. Premature Ovarian Insufficiency - an update on recent advances in understanding and management. F1000 Res. 2017; 6: 2069. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5710309/
- Fenton AJ. Premature ovarian insufficiency: Pathogenesis and management. J Midlife Health. 2015; 6: 147-153. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4743275/
- Hodis HN, Mack WJ, Henderson VW, Shoupe M, Budoff MJ, et al. Vascular Effects of Early versus Late Postmenopausal Treatment with Estradiol. N Engl J Med. 2016; 374: 1221-1231. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4921205/
- Mohammed K, Dabrh AMA, Benkhadra K, Al Nofal A, Leon BGC, et al. Oral vs Transdermal Estrogen Therapy and Vascular Events: A Systematic Review and Meta-Analysis. J Clin Endocrinol Metab. 2015; 100: 4012-4020. PubMed: https://pubmed.ncbi.nlm.nih.gov/26544651/
- Ge W, Li L, Dyce PW, De Felici M, Shen W. Establishment and depletion of the ovarian reserve: physiology and impact of environmental chemicals. Cell Mol Life Sci. 2019; 76: 1729-1746. PubMed: https://pubmed.ncbi.nlm.nih.gov/30810760/
- Vujovic S, Brincat M, Erel T, Gambacciani M, Lambrinoudaki I, et al. EMAS position statement: Managing women with premature ovarian failure. Maturitas. 2010; 67: 91-93. PubMed: https://pubmed.ncbi.nlm.nih.gov/20605383/
- Popat VB, Calis KA, Vanderhoof VH, Cizza G, Reynolds JC, et al. Bone mineral density in estrogen-deficient young women. J Clin Endocrinol Metab, 2009. 94: 2277-2283. PubMed: https://pubmed.ncbi.nlm.nih.gov/19401379/
- Schmidt J, Cardoso GMP, Ross JL, Haq N, Rubinow DR, et al. Shyness, social anxiety, and impaired self-esteem in Turner syndrome and premature ovarian failure. JAMA. 2006; 295: 1374-1376. PubMed: https://pubmed.ncbi.nlm.nih.gov/16551707/