
More Information
Submitted: May 05, 2021 | Approved: May 19, 2021 | Published: May 20, 2021
How to cite this article: Akhrif M, Sabib M, Rouas L, Meskini T, Mouane N. A rare cause of neonatal diarrhoea: Microvillositary inclusion disease: about a case report. J Adv Pediatr Child Health. 2021; 4: 053-056.
DOI: 10.29328/journal.japch.1001033
Copyright License: © 2021 Akhrif M, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

A rare cause of neonatal diarrhoea: Microvillositary inclusion disease: about a case report
M Akhrif1,2*, M Sabib1, L Rouas3, T Meskini1 and N Mouane1
1Department of Pediatric Hepatology Gastroenterology and Nutrition, Rabat Children’s Hospital, Morocco
2Faculty of Medicine and Pharmacy, Morocco
3Pathological Anatomy Laboratory, Ibn Sina Hospital, Rabat, Morocco
*Address for Correspondence: M Akhrif, Department of Pediatric Hepatology Gastroenterology and Nutrition, Rabat Children’s Hospital, Morocco, Tel: 212.0642536361; Email: [email protected]
Microvillositary inclusion disease also known as microvillositary atrophy is a rare congenital enteropathy containing a border abnormality in the brushes of enterocytes, manifesting as severe rebellious diarrhea in newborns and infants. It was first described in 1978 by Davidson, et al. The autosomal recessive mode of transmission is suggested because of the frequency of familial cases and inbreeding. Histopathology plays an essential role in establishing the diagnosis. In 2008, a common mutation was identified in most of the patients studied in the MYO5B gene that codes for the Myosin Vb protein, which helped in understanding the etiopathogeny of this pathology poorly described in the literature. The prognosis for this pathology is extremely bleak, requiring total parenteral nutrition for child survival. Intestinal transplantation is for the moment the only long-term solution.
Materials and methods: We report the case of an infant aged 6 months, with no perinatal antecedent. There is 1st degree consanguinity, the mother has a history of deaths in younger siblings in undetermined circumstances. Who since the age of 3 days presents profuse liquid diarrhoea with malnutrition, dehydration and enormous abdominal distension? Several diagnoses were suspected before the jejune biopsy was carried out, which led to the diagnosis of a microvilliositary inclusion disease.
The aim of our work is to highlight the rarest cause of neonatal rebel diarrhoea and to know how to include it among other differential diagnoses.
Microvillous Inclusion Disease (MVID) belongs to the group of refractory diarrhoea in the youngest infant that causes intestinal failure during the first days or months of life. It is caused by a severe congenital alteration of the intestinal epithelium resulting in massive watery diarrhoea and permanent malabsorption, usually leading to dependence on total parenteral nutrition (TPN) [1]. Although the disease is rare and only about 200 cases of IDMC have been reported, it is more common in countries where consanguineous marriages are common [2]. MVID is characterized by defective transport of plasma membrane proteins to the apical brush border, due to mutations of MYO5B gene on the chromosome 18q21, encoding myosin Vb motor and a small GTP-binding protein, Rab8 [3]. It is an entity that must be remembered in the case of neonatal diarrhoea appearing during the first 72 hours of life, without an underlying infectious or metabolic cause [4].
We report the case of a male infant, 6 months old, only son of his family; he was born at term, with no antenatal or perinatal incidences, with a birth weight of 3 kg. He was initially breastfed, and later mixed breastfeeding. There is 1st degree consanguinity, the mother has a history of deaths in younger siblings in undetermined circumstances. Admitted for severe dehydration and malnutrition.
The history of his illness goes back to the neonatal period; at the third day of life, with profuse liquid diarrhea at the rate of 10 to 12 stool per day associated with abdominal bloating and weight stagnation.
There was no fever, no skin or respiratory signs, no jaundice and no recurrent infection. He also had a few sub-occlusive episodes with no surgical indication, which responded well to nursing. Diarrhea persists during digestive rest and worsens during feeding.
Clinical examination revealed an apathetic infant with severe denutrition and signs of dehydration associated with significant abdominal distension (Figure 1) with a staturoponderal delay: Weight: 4.45 kg (-3DS), Height: 54 cm (-3DS), PC: 43.5 cm (-3DS), Brachial perimeter: 9 cm. The rest of the clinical examination is without any particularities. Notably, no dysmophysis, no hepatomegaly or splenomegaly, a normal pleuropulmonary examination, a normal neurological examination apart from a slight axial hypotonia.
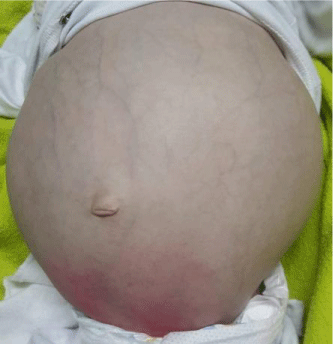
Figure 1: Picture of our patient shows the enormous abdominal distension.
In the face of these clinical signs, several diagnoses of chronic diarrhoea were suspected, so the biological and radiological investigations were aimed at informing or confirming the differential diagnoses.
The initial biological tests revealed a hypernatremia at 183 mEq/l.
After rehydration, the natremia has corrected, however, a renal tubular acidosis is present with alkaline reserve at 11 mEq/l, blood urea at 1.49 g/l, creatinine at 16.2, calcemia at 128 mg/l, vitamin D: 17 ng/ml, parathormone at 12.10 pg/ml (N). The urinary test showed significant crystalluria, calciuria at 610 mg/l, urinary urea at 2.14 g/l.
It was not possible to perform a stool ionogram.
Infectious test is negative. HIV or congenital immune deficiency test is negative.
Abdominal ultrasound revealed bilateral nephrocalcinosis grade 1.
Exploration of subocclusive episodes with opacification and rectal biopsy is without abnormalities.
The fecal elastase dosage is reduced to: 26 ug/g of stool: showed an exocrine pancreatic insufficiency, so the infant was supplemented with pancreatic extract, however immunoreactive trypsin is normal.
Upper intestinal endoscopy with jejunal biopsy led to the diagnosis of microvillositid atrophy (Figures 2,3).
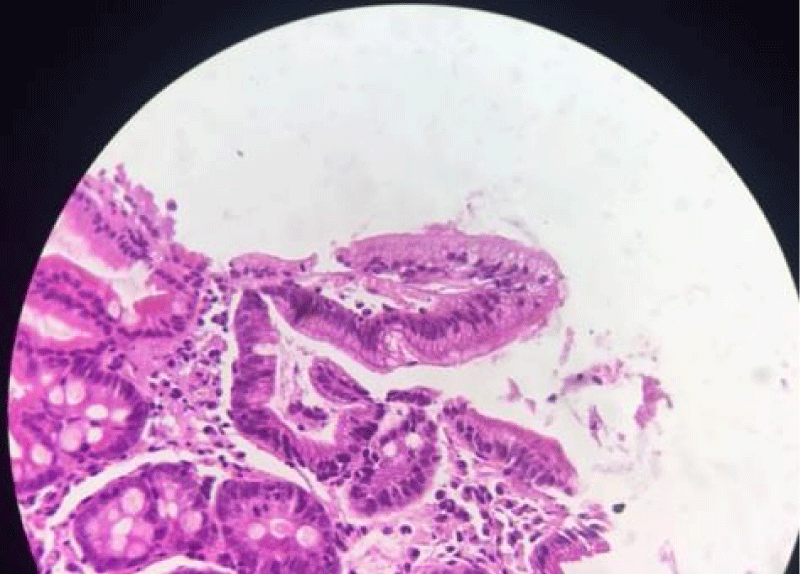
Figure 2: Hematoxylibe eosin; x 20 intestinal villosity with microvacuolar aspect of the apical pole of the enterocytes and a non visible brush border.
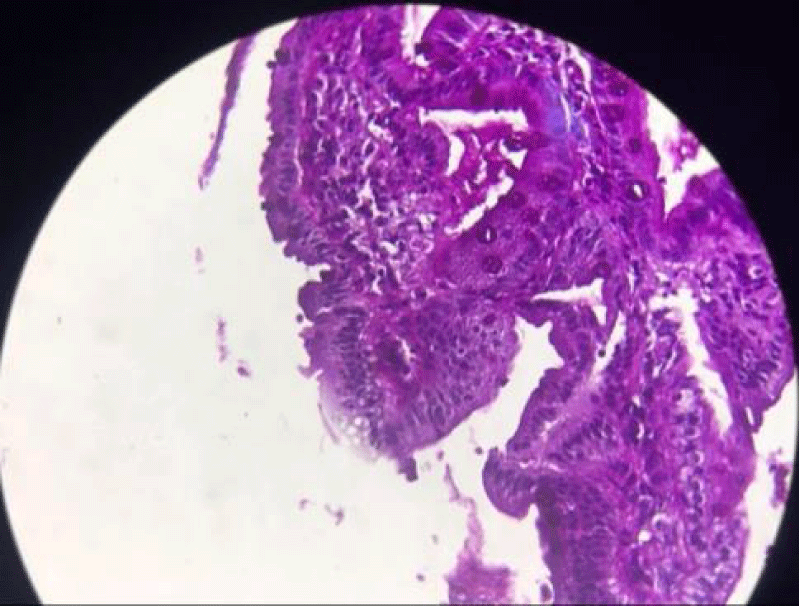
Figure 3: PAS, positive marriage of the apical pole of the entrrocytes.
A genetic study is requested but not carried out due to lack of means.
The infant was supplemented with fat-soluble vitamins, with continuous enteral feeding with hydrolyzed milk, noted that digestive rest did not show a significant reduction in the number of stools.
Microvillous inclusion disease or microvillous atrophy (MVA) is a congenital and constitutive disorder of intestinal epithelial cells. Its prevalence is currently estimated at < 1:1,000,000 and there are few known cases in the world [1]. Severe refractory diarrhoea in the neonatal period is rare. However, among this group of exceptional diseases, MVID is the most common cause of neonatal diarrhoea [5]. There is a slight preponderance of sex female. Pregnancy and childbirth in general occur without associated pathology, it is like the case of our patient, he had no ante-natal anomaly, although oligohydroamnios have been described in some cases [1].
Previous studies in patients with MVID have shown a high stool volume (150-300 ml/kg/day) with a remarkably high sodium content (about 100 mmol/L) [6]. The diarrhoea present during MVID is considered to be non-osmotic in nature (i.e. the difference between faecal ions is less than 100 mOsm) and is persistent even when the patient is unfed [7]. This type of diarrhoea is classified as electrolyte transport diarrhoea, caused by mechanisms involving net secretion of anions (chloride, bicarbonate or potassium) and/or net inhibition of sodium or chloride absorption [8]. Steatorrhea and impaired glucose absorption have also been reported in patients with MVID [9]. Following diarrhoea, children suffer from dehydration and metabolic decompensation. Hypovolemia causes ischaemia with developmental delay. They may also have impaired kidney function and calcinosis [4]. As in the case of our patient with psychomotor retardation, hydroeletrolytic disorders and biateral nephrocalcinosis.
Histopathology plays an essential role in establishing the diagnosis, a combination of light and electron microscopy of duodenum, jejunum and colon samples is used for diagnosis. In the first, with staining with haematoxylin and eosin, hypoplastic but variable atrophy of the villi and accumulation of acid-stained secretory granules is observed Schiff’s Periodical (PAS) positive in cytoplasm apical of enterocytes, without inflammatory infiltration [10]. The MVID can be divided into three types:
- Early MVID with complete loss of protein from microvilli in the villi and crypts.
- Late-onset MVID, where microvilli proteins are absent at the ends of the villi and on the sides, but normal microvilli are present at the base of the villi and crypts. These patients become symptomatic as early as the second month after birth, and the symptoms are less severe.
- Atypical MVID, where microvilli proteins such as vinculin and syntaxin are absent or defective only at the crypts, but where normal microvilli are present on the surface of the villi.
The crypts remain intact. Electronic microscopy is used to recognise pathognomonic changes that are intracytoplasmic vacuoles with microvilli included and absence or scarcity of microvilli on the luminal edge. An immunoreactive stain can also be performed to recognise a neutral CD-10 peptidase which in this disease is located within the cytoplasm of the enterocyte in contrast to the normal intestine or other intestinal pathologies in which is usually demonstrated on the surface [11]. The identification of genetic mutations linked to trafficking pathways in MVID has paved the way for further research to better understand this complex and difficult enteropathy. The main mutation observed in MVID patients is in the MYO5B gene, the key molecular motor gene that regulates the trafficking of important proteins to the brush border of the gut [12].
Within the differential diagnostics there are consider other causes of intractable diarrhoea in infants such as congenital carrier defects, epithelial dysplasia or enteropathy in plume, autoimmune enteropathy, IPEX syndrome, as well as allergies, infections and enteropathies post-infection [13]. In our patient, biological investigations have eliminated differential diagnoses.
The MYO5B gene is expressed in all epithelial tissues, but the most significant phenotype is expressed in the intestine. Nevertheless, several extra-intestinal pathologies have also been reported in other tissues. In this regard, the pathologies identified include Fanconi’s kidney syndrome, cholestasis, haematuria and pneumonia. [14].
The prognosis for this disease is most often severe. The life expectancy of these patients is reduced, with a one-year survival rate of 25%. The majority of patients die due to complications from eating prolonged parenteral (sepsis, hepatocellular insufficiency, metabolic disorders) [4].
The only therapeutic option is TPN and intestinal rest and isolated intestinal transplantation or combined liver/small intestine transplantation. The evolution of our patient is marked by episodes of dehydration and severe under nutrition, as intestinal transplantation is not a feasible therapeutic option in our country [15].
Microvillous atrophy is a rare intestinal disorder that manifests itself as severe rebellious diarrhoea in newborns and infants. It is an entity that must be considered in the face of rebellious neonatal diarrhoea to avoid reaching the final stage of complications, even if the only therapeutic option is intestinal transplantation, which is not accessible to all patients.
- Schoen K, Puchi A, González I, Torres MT, Espinosa R, et al. Enfermedad por inclusión microvellositaria como causa de diarrea congénita severa. Caso clínico [Microvillous inclusion disease as a cause of severe congenital diarrhea. Case report]. Rev Chil Pediatr. 2017; 88: 662-667.
- Phulware RH, Gahlot GPS, Malik R, Gupta SD, Das P. Microvillous Inclusion Disease as a Cause of Protracted Diarrhea. Indian J Pediatr. 2019; 86: 854-856. PubMed: https://pubmed.ncbi.nlm.nih.gov/31049800/
- Mahjoub F, Niknejad S, Sadeghian M, Abdirad A. Congenital microvillous atrophy, report of two consecutive siblings with complete histologic, immunohistochemical and detailed electron microscopic studies, first report from Iran. Open J Pathol. 2014; 4: 64–67.
- Guellouz N, Zekri S, Youssef A, Zghald D, Ben Hariz M, et al. Maladie des inclusions microvillositaires à manifestation anténatale : à propos de trois observations familiales. J de Pédiatrie et de Puériculture. 2011; 24: 72-76.
- Khubchandani SR, Vohra P, Chitale AR, Sidana P. Microvillous inclusion disease--an ultrastructural diagnosis: with a review of the literature. Ultrastruct Pathol. 2011; 35: 87-91.
- Oktavia Sari Y, Bahari MB, Ibrahim B. Clinical review of total parenteral nutrition use among pediatric: Critics and outcomes. Int J Pharm Life Sci. 2013; 4.
- Canani RB, Terrin G: Recent progress in congenital diarrheal disorders. Curr Gastroenterol Rep. 2011; 13: 257–264. PubMed: https://pubmed.ncbi.nlm.nih.gov/21494839/
- Thiagarajah JR, Kamin DS, Acra S, Goldsmith JD, Roland JT, et al. Advances in Evaluation of Chronic Diarrhea in Infants. Gastroenterology. 2018; 154: 2045-2059.e6. PubMed: https://pubmed.ncbi.nlm.nih.gov/29654747/
- Iancu TC, Manov I. Ultrastructural aspects of enterocyte defects in infancy and childhood. Ultrastruct Pathol. 2010; 34: 117–125. PubMed: https://pubmed.ncbi.nlm.nih.gov/20455660/
- Phillips AD, Szafransky M, Man LY, Wall W. Periodic Acid Schiff Staining Abnormality in Microvillous Atrophy: Photometric and Ultrastructural Studies. J Pediatr Gastroenterol Nutr. 2000; 30: 34-42. PubMed: https://pubmed.ncbi.nlm.nih.gov/10630437/
- Khubchandani SR, Vohra P, Chitale AR, Sidana P. Microvillus inclusion disease-An Ultrastructural Diagnosis: with a Review of the literatura. Ultrastruct Pathol. 2011; 35: 87-91. PubMed: https://pubmed.ncbi.nlm.nih.gov/21299349/
- Jayawardena D, Alrefai WA, Dudeja PK, Gill RK. Recent advances in understanding and managing malabsorption: focus on microvillus inclusion disease. F1000Res. 2019; 8: F1000. PubMed: https://pubmed.ncbi.nlm.nih.gov/31824659/
- Volonaki E, Sebire N, Borrelli O, Lindley KJ, Elawad M, et al. Gastrointestinal Endoscopy and Mucosal Biopsy in the First Year of Life: Indications and Outcome. J Pediatr Gastroenterol Nutr. 2012; 55: 62-65. PubMed: https://pubmed.ncbi.nlm.nih.gov/22210413/
- Golachowska MR, van Dael CM, Keuning H, Karrenbeld A, Hoekstra D, et al. MYO5B mutations in patients with microvillus inclusion disease presenting with transient renal Fanconi syndrome. J Pediatr Gastroenterol Nutr. 2012; 54: 491–498. PubMed: https://pubmed.ncbi.nlm.nih.gov/22441677/
- Bulut O, Ahishali B, Gulluoglu M, Arslanoglu S. A Case Study of Intractable Diarrhea Due to Neonatal Microvillous Inclusion Disease, Fetal Pediatr Pathol. 2017; 36: 340-343. PubMed: https://pubmed.ncbi.nlm.nih.gov/28707991/