
More Information
Submitted: October 25, 2022 | Approved: November 22, 2022 | Published: November 23, 2022
How to cite this article: Bilge S, Özcan N, Özcanyüz DG, Mert GG, İncecik F, et al. Atypic subacute sclerosing panensefalitis in a six-year-old male. J Adv Pediatr Child Health. 2022; 5: 039-041.
DOI: 10.29328/journal.japch.1001051
Copyright License: © 2022 Bilge S, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Subacute sclerosing panencephalitis; Myoclonic seizure; Atypical course; Ataxia

Atypic subacute sclerosing panensefalitis in a six-year-old male
Serap Bilge*, Neslihan Özcan, Duygu Güner Özcanyüz, Gülen Gül Mert, Faruk İncecik and M Özlem Hergüner
Department of Pediatric Neurology, Faculty of Medicine, Cukurova University, Adana, Turkey
*Address for Correspondence: Serap Bilge, Department of Pediatric Neurology, Faculty of Medicine, Cukurova University, Adana, Turkey, Email: [email protected]
Subacute sclerosing panencephalitis is a rare, slow, and insidious neurodegenerative disease caused by measles. This disease mostly has a classic course. However, sometimes it can be presented with atypical manifestations. In this paper, we aim to present a six years old male patient that was hospitalized due to seizures and ataxia. Cerebral magnetic resonance imaging was normal on the first day of admission, but within a few days, the patient started to be apathetic. On the seventh day, magnetic resonance imaging showed hyperintense lesions in the thalamic, brainstem, and periventricular areas. Periodic epileptiform discharges were detected in the repeated electroencephalogram. Investigations from the cerebrospinal fluid showed markedly elevated measles virus IgG at 230U/ml consistent with the diagnosis of SSPE which should always be ruled out when a patient comes in with uncontrollable seizures, ataxia and apathy.
Subacute Sclerosing Panencephalitis (SSPE) is a rare, slow, and insidious neurodegenerative disease caused by measles. The disease can develop due to the reactivation of the measles virus or an improper immune response to this virus. Behavioral problems, personality changes, seizures, cognitive deterioration, and pyramidal, or extrapyramidal findings are the main features of the clinic. However, SSPE can also follow a fulminant course [1].
Dyken’s criteria are based on the diagnosis which includes two major and four minor criteria. Raised anti-measles antibody titers in cerebrospinal fluid (CSF) greater than or equal to 1:4 or ratio greater than or equal to 1:256 in serum, typical or atypical clinical history are major criteria. Minor criteria include characteristic electroencephalographic findings that include periodic, generalized, bilaterally synchronous and symmetrical high-amplitude slow waves that recur at regular intervals of 5 - 15 seconds called periodic slow-wave complexes, CSF globulin levels greater than 20% of the total CSF protein, characteristic histopathological findings on brain biopsy including inflammatory changes and specialized molecular diagnostic test to identify wild-type measles virus mutated genome. Usually, two major criteria plus one minor criterion are required [1]. Here we reported a case of SSPE that showed interesting clinical courses and radiologic features.
A six years old male patient was hospitalized due to refractory seizures and progressive gait. The patient was born to consanguineous parents, and antenatal and perinatal follow-up reported no insults. There was no family history of epilepsy or neurological diseases. At the age of one, seizures started and they were taken under control with levetiracetam. Metabolic screenings and cerebral Magnetic Resonance Imaging (MRI) performed at that time were normal. He was admitted to our clinic due to frequent seizures and ataxia. On physical examination, the patient was conscious, and cranial nerve and motor examination were normal but ataxia of the trunk and lower extremities were noticed. The epileptiform abnormalities with multifocal myoclonic features were detected on EEG. Antiepileptic treatment was adjusted. Metabolic screenings, and viral, autoimmune encephalitis panel were normal. Only high blood measles IgG (+) was detected. On the first day of hospitalization, the cerebral MRI didn’t show any abnormalities, but the patient started to be apathetic within a few days, on the 7th day, MRI showed hyperintense lesions in the T2A sequences in the area of the brainstem, thalamic region, periventricular, and caudate nucleus (Figure 1). Periodic epileptiform discharges were detected in the repeated EEG (Figure 2). Investigations from the cerebrospinal fluid (CSF) showed markedly elevated measles virus IgG at 230 U/ml (N: 0 - 25) consistent with the diagnosis of SSPE. On the 35th day of hospitalization, the seizures were taken under control using clobazam and carbamazepine and the patient was discharged in a vegetative state.
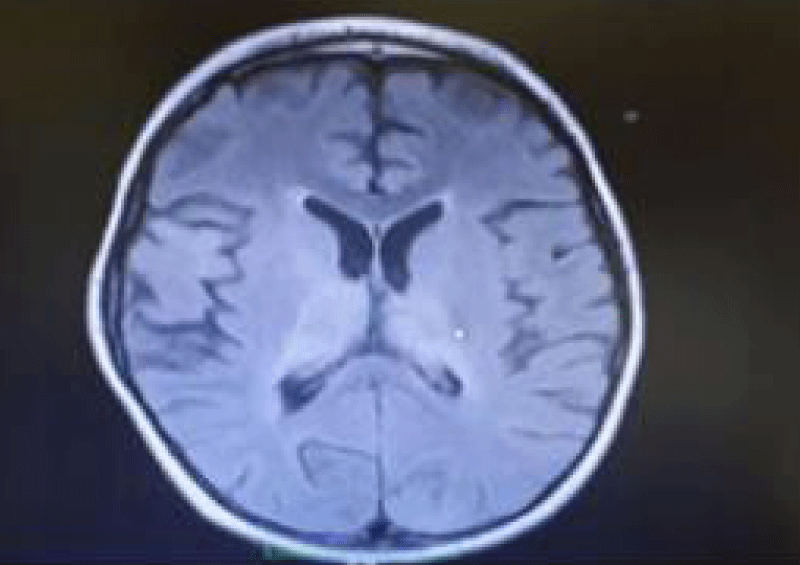
Figure 1: Hyperintensities on T2-weighted images in the periventricular white matter cerebral cortex and basal ganglia, thalamus in a six years old boy with SSPE.
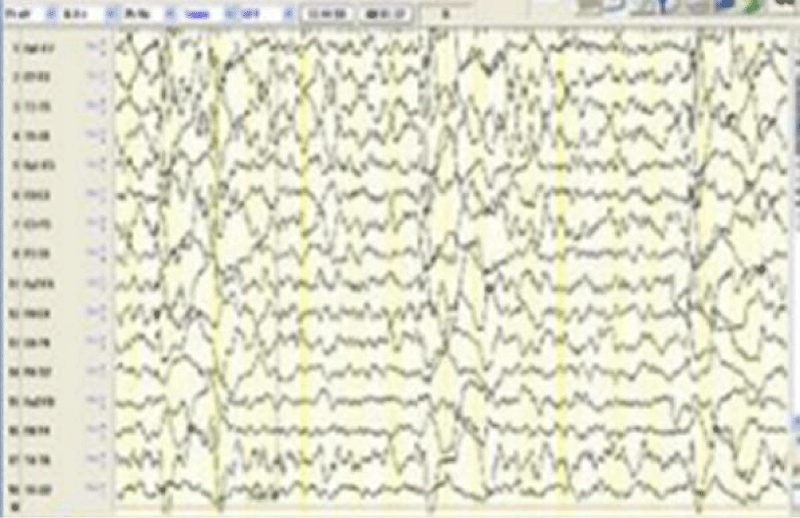
Figure 2: EEG shows the periodic discharges in six years old patients with SSPE.
SSPE is a progressive neurological disease that usually occurs during childhood and early adolescence. SSPE can be presented in two clinical forms; typical classic course and atypical. The classic one has 4 stages; (First stage) Behavioral disorders and intellectual regression, (Second stage) Involuntary movements, (Third stage) Severe pyramidal-extrapyramidal findings, (Fouth stage) Chronic vegetative period, and death. Generally, all phases last between months and years [1]. Atypical features such as acute encephalitis [2], dysarthria, ataxia [3,4], abrupt vision loss [5]. Epilepsia partialis continua [6], focal deficits, and asymmetric myoclonus [7] have been reported. SSPE can be manifested as myoclonic-astatic epilepsy. An Italian girl died within four months after the onset of the disease was reported. Unfortunately, delays in the diagnosis and treatment of SSPE can occur mostly in patients with an atypical course [8]. Seizure disorders poorly controlled with medication could be the only manifestation of atypical cases. Catching the measles virus before the age of two, increased viral virulence, and coinfection with other viruses are risk factors for a more fulminant, atypical course [9]. Another case of SSPE that presented with ataxia and right hemiplegia with no visual symptoms seizures or slow generalized myoclonus has been reported. The asymmetrical slowing on the initial EEG only suggested a left hemispheric insult [4]. Our patient who was diagnosed with epilepsy earlier had uncontrollable seizures on admission and developed progressive ataxia. The first EEG showed multifocal discharges which urged us to think of the diseases that cause progressive myoclonic seizures. Our patient became apathetic on the third day the possibility of Nonconvulsive Status Epilepticus (NCSE) was suspected. Therefore there was a need for a second EEG. Continuous EEG was recorded on the third day which showed periodic slow-wave discharges that were consistent with the diagnosis of SSPE. In this article, we present an atypical case to highlight that SSPE is still a cause of encephalopathy that should be always kept in mind when investigating a patient with neurological symptoms.
Neuroimaging can be beneficial and supportive but it is not characteristic and could be normal in the early stages of SSPE. MRI of the brain may show decreased gray matter volume, especially within the frontotemporal cortex, amygdala and cingulate gyrus during the early stages, Hyperintensities on T2-weighted images in the periventricular white matter of cerebral cortex, brainstem, and basal ganglia, may develop. As the disease progresses. MRI could also reveal diffuse cortical atrophy [10]. Our patient’s MRI was normal on admission but within 7 days it revealed hyperintensities in the thalamic, brainstem, and periventricular area.
The major part of the therapeutic strategy is supportive and symptomatic treatment. Ant iviral drugs and immuno-modulators are used in the treatment, although there is no standard treatment for SSPE. The most commonly used drugs in clinical practice are Inosine pranobex, interferon alfa, ribavirin, and lamivudine. Steroids and intravenous immunoglobulin are no longer recommended. Antiepileptics and the ketogenic diet are supportive in the treatment of SSPE [1].
Atypical presentations of SSPE should be always kept in mind and ruled out in every patient who comes with uncontrollable seizures, ataxia, and apathy to prevent further delays of the diagnosis.
Informed consent statement: The parents or the child’s legal guardians provided informed written consent before study enrollment.
STROBE statement: The authors have read the STROBE Statement - a checklist of items, and the manuscript was prepared and revised according to the STROBE Statement - a checklist of items.
- Jabbour J, Duenas D, Modlin J. SSPE-clinical staging, course, and frequency. Arch Neurol. 1975;32(7):493–494.
- Komur M, Arslankoylu AE, Okuyaz C, Kuyucu N. Atypical clinical course subacute sclerosing panencephalitis presenting as acute Encephalitis. J Pediatr Neurosci. 2012 May;7(2):120-2. doi: 10.4103/1817-1745.102574. PMID: 23248691; PMCID: PMC3519069.
- Hergüner MO, Altunbaşak S, Baytok V. Patients with acute, fulminant form of SSPE. Turk J Pediatr. 2007 Oct-Dec;49(4):422-5. PMID: 18246746.
- Kandadai RM, Yada P, Uppin MS, Jabeen SA, Cherian A, Kanikannan MA, Borgohain R, Challa S. Fulminant subacute sclerosing panencephalitis presenting with acute ataxia and hemiparesis in a 15-year-old boy. J Clin Neurol. 2014 Oct;10(4):354-7. doi: 10.3988/jcn.2014.10.4.354. Epub 2014 Oct 6. PMID: 25324886; PMCID: PMC4198718.
- Ekici B, Calişkan M, Tatli B, Aydinli N, Ozmen M. Rapidly progressive subacute sclerosing panencephalitis presenting with acute loss of vision. Acta Neurol Belg. 2011 Dec;111(4):325-7. PMID: 22368974.
- Kravljanac R, Jovic N, Djuric M, Nikolic L. Epilepsia partialis continua in children with fulminant subacute sclerosing panencephalitis. Neurol Sci. 2011 Dec;32(6):1007-12. doi: 10.1007/s10072-010-0442-y. Epub 2010 Oct 30. PMID: 21052755.
- Shivji ZM, Al-Zahrani IS, Al-Said YA, Jan MM. Subacute sclerosing panencephalitis presenting with unilateral periodic myoclonic jerks. Can J Neurol Sci. 2003 Nov;30(4):384-7. doi: 10.1017/s0317167100003127. PMID: 14672273.
- Magurano F, Marella GL, Marchi A, Filia A, Marsella LT, Potenza S, Massa R, Bucci P, Baggieri M, Nicoletti L. A case of fulminant subacute sclerosing panencephalitis presenting with acute myoclonic-astatic epilepsy. Ann Ist Super Sanita. 2017 Apr-Jun;53(2):167-169. doi: 10.4415/ANN_17_02_15. PMID: 28617265.
- Jafri SK, Kumar R, Ibrahim SH. Subacute sclerosing panencephalitis - current perspectives. Pediatric Health Med Ther. 2018 Jun 26;9:67-71. doi: 10.2147/PHMT.S126293. PMID: 29985487; PMCID: PMC6027681.
- Akdal G, Baklan B, Cakmakçi H, Kovanlikaya A. MRI follow-up of basal ganglia involvement in subacute sclerosing panencephalitis. Pediatr Neurol. 2001 May;24(5):393-5. doi: 10.1016/s0887-8994(01)00267-3. PMID: 11516619.