
More Information
Submitted: December 27, 2023 | Approved: January 17, 2024 | Published: January 18, 2024
How to cite this article: Al-essi MA, Binkhamis LS, Aljohani SM, Alzahrani NM. Delayed Diagnosis of Early-onset Sarcoidosis: A Case Report and Literature Review. J Adv Pediatr Child Health. 2024; 7: 001-006.
DOI: 10.29328/journal.japch.1001061
Copyright License: © 2024 Al-essi MA, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Early-onset sarcoidosis; Childhood sarcoidosis; Arthritis; Granulomatous; Sarcoidosis; Blau syndrome
Abbreviations: CS: Classic Sarcoidosis; EOS: Early-Onset Sarcoidosis; NOD2: Nucleotide-binding Oligomerization Domain-containing protein 2; CARD15: Caspase Recruitment Domain-containing protein 15; JIA: Juvenile Idiopathic Arthritis; MRI: Magnetic Resonance Imaging; WES: Whole Exome Sequencing; ACE: Angiotensin-Converting Enzyme; WASOG: World Association of Sarcoidosis and other Granulomatous Disorders

Delayed Diagnosis of Early-onset Sarcoidosis: A Case Report and Literature Review
Mutibah Ali Al-essi1*, Lujain Salah Binkhamis2, Samah Mohammed Aljohani3 and Nora Mohammad Alzahrani4
1Pediatric Rheumatologist, King Fahd Hospital of University, Alkhobar, Saudi Arabia
2Department of General Surgery, King Fahd Hospital of University, Alkhobar, Saudi Arabia
3Department of Dermatology, King Fahd Hospital of University, Alkhobar, Saudi Arabia
4Medical Intern at Imam Abdulrahman Bin Faisal University, King Fahd Hospital of University, Alkhobar, Saudi Arabia
*Address for Correspondence: Mutibah Ali Al-essi (MA), Pediatric Rheumatologist, King Fahd Hospital of University, Alkhobar, Saudi Arabia, Email: [email protected]
Background: Early-onset sarcoidosis is a rare systemic inflammatory granulomatous disease, distinguished by onset before the age of 4 and notably lacking pulmonary involvement. Unfortunately, the condition often shows clinical features similar to juvenile idiopathic arthritis, resulting in the misdiagnosis of numerous patients. This case report delves into the challenges associated with the delayed diagnosis of early-onset sarcoidosis, with a particular focus on the diagnostic methods employed to address this delayed recognition.
Case presentation: A 15-year-old girl presented with a history of recurrent fever since infancy, accompanied by rash, arthritis, and joint deformity. Previously misdiagnosed with juvenile idiopathic arthritis, she underwent management with steroids and methotrexate, yielding no improvement. The diagnosis of early-onset sarcoidosis was made during adolescence based on serial examinations, comprehensive laboratory and radiological evaluations, and subsequent histopathology findings. Presently, the patient is receiving treatment with low-dose steroids and biologic therapy (Tocilizumab) and experiencing no disease progression.
Conclusion: This case report underscores the importance of considering early-onset sarcoidosis in the differential diagnosis of pediatric patients exhibiting persistent arthritis from an early age. Early detection and treatment are crucial in averting complications and enhancing the overall quality of life.
Sarcoidosis is a rare autoinflammatory disease that affects multiple body organs, with its underlying etiology remaining unknown. The identification of this condition relies on the presence of noncaseating epithelioid cell granulomas observed in biopsy specimens from different body organs [1]. The diagnosis of sarcoidosis in the pediatric age group can be challenging due to its diverse presentation. In pediatrics, two different types of sarcoidosis have been reported: Classic Sarcoidosis (CS) and Early-Onset Sarcoidosis (EOS). Classic sarcoidosis usually affects older children (> 5 years old) and exhibits clinical manifestations similar to those observed in adults, characterized by pulmonary, lymph node, and ocular involvement. Conversely, early-onset sarcoidosis, a rarer occurrence, manifests in younger children with no apparent pulmonary involvement [2].
EOS manifests as a triad of arthritis, uveitis, and cutaneous manifestations, representing the sporadic form of autoinflammatory systemic granulomatous diseases. Its counterpart, Blau syndrome, exists as a familial form inherited in an autosomal dominant manner, sharing a clinical presentation similar to EOS but lacking familial history. Typically affecting young children before the age of 4 years, its onset primarily involves cutaneous and articular symptoms, distinguishing it from the typical lung disease observed in older children and adults [3].
The disease has been associated with a mutation in the nucleotide-binding oligomerization domain-containing protein 2 (NOD2), also termed caspase recruitment domain-containing protein 15 (CARD15), which is mapped on chromosome 16q12. This mutation is responsible for the development of granulomatous inflammation observed in EOS/Blau syndrome [4]. This NOD2 protein plays an important role in the inflammatory response through microbe-associated molecular pattern recognition. Among the most frequently detected NOD2 mutations are R334W and R334Q [5].
For EOS presenting with multisystem involvement, corticosteroids stand as the primary therapy of choice. However, it is important to tailor the duration and dosage of the therapy to individual needs. Methotrexate and biologic agents, such as Tocilizumab, demonstrate efficacy and have steroid-sparing properties [6].
EOS and Blau syndrome are rare granulomatous autoinflammatory diseases affecting young children and are caused by (NOD2/CARD15) mutations. Affected children typically exhibit cutaneous manifestations, joint symptoms, and uveitis. Given the non-specific clinical features and the lack of a specific test, identifying sarcoidosis in the pediatric population can be challenging [7]. Consequently, it usually gets misdiagnosed as Juvenile Idiopathic Arthritis (JIA), as was the case with our patient.
Here, we present the case of a 15-year-old girl recently diagnosed with early-onset sarcoidosis, showing manifestations of delayed diagnosis and consequential adverse effects stemming from prolonged corticosteroid therapy.
A 15-year-old girl visited King Fahd Hospital University in October 2021 with a history of recurrent daily low-grade fever since the age of 1 year, joint deformity accompanied by arthralgia, and generalized bone pain. In her medical history, she was diagnosed with polyarticular juvenile idiopathic arthritis and bilateral anterior uveitis, leading to management with steroids and methotrexate; however, no disease remission was achieved. There were no notable instances of autoimmune diseases in her family history.
A series of examinations conducted upon admission revealed her weight and height to be below the third percentile, indicative of short stature. Vital signs showed tachycardia, with a heart rate of 130 bpm, and a low-grade fever, with a temperature of 38.5. Physical examination revealed transparent skin with visible veins in the abdominal region. Musculoskeletal examination revealed bilaterally swollen wrists and ankles with severe movement restriction, along with multiple symmetrically distributed tiny erythematous scaly xerotic papules on the trunk, upper limbs, and lower limbs (Figure 1). Furthermore, bilateral flexion deformities of the interphalangeal joints of the ring and middle fingers were observed (Figure 2). Additionally, there was restricted dorsiflexion of the ankle joints due to tightness in both the gastrocnemius and soleus muscles. Examination of the back showed lumber kyphosis with prominent spinous processes (Figure 3), and tenderness was detected at the L4 level. A slit-lamp test showed no signs of active uveitis. Other aspects of the physical examination were unremarkable.
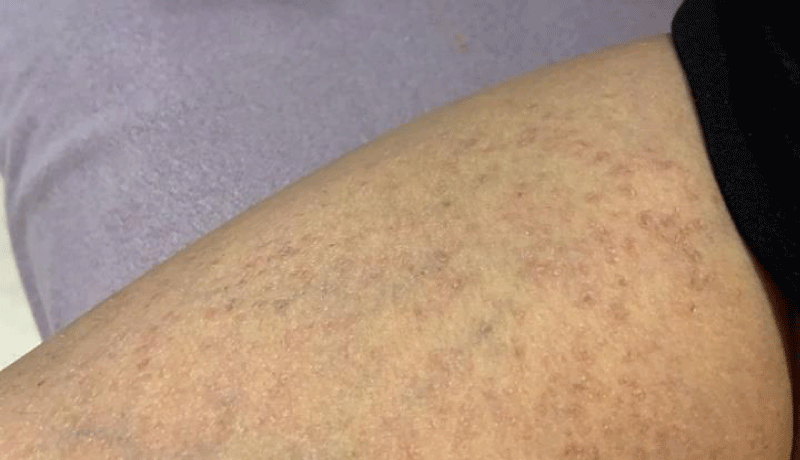
Figure 1: Multiple symmetrically distributed tiny erythematous scaly xerotic papules on the thigh.
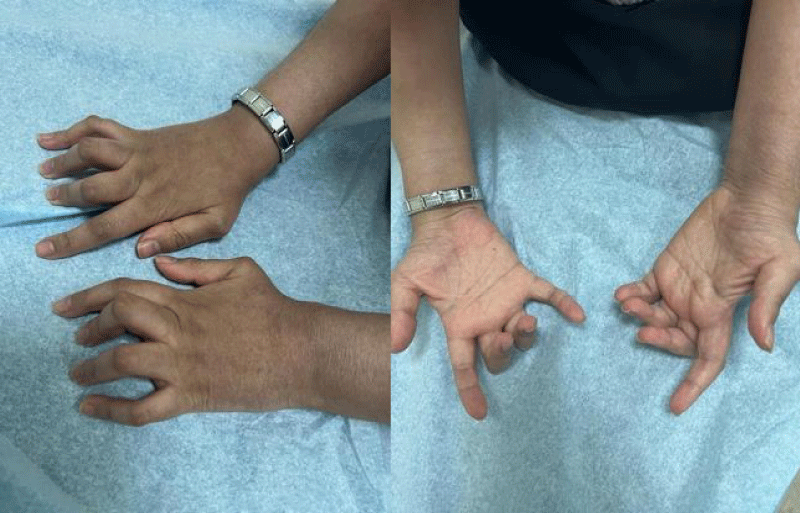
Figure 2: Bilateral flexion of the interphalangeal joints of the ring and middle finger.
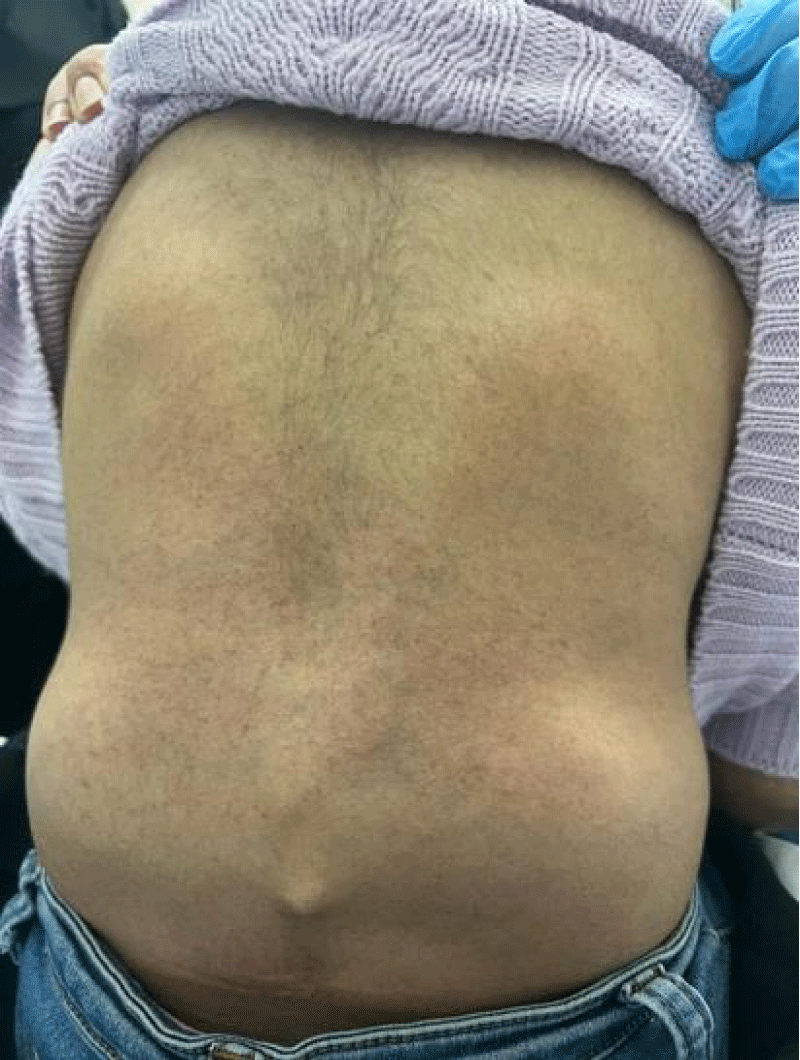
Figure 3: Lumbar kyphosis with prominent spinous processes.
To monitor the period of admission, a number of radiological, laboratory, and histopathological investigations were conducted. A hand X-ray was performed and revealed a generalized reduction in bone density accompanied by periarticular osteoporotic changes. Notably, a pathological fracture was seen in the distal left radius (Figure 4). Additionally, a chest X-ray exhibited degenerative changes in both shoulder joints (Figure 5).
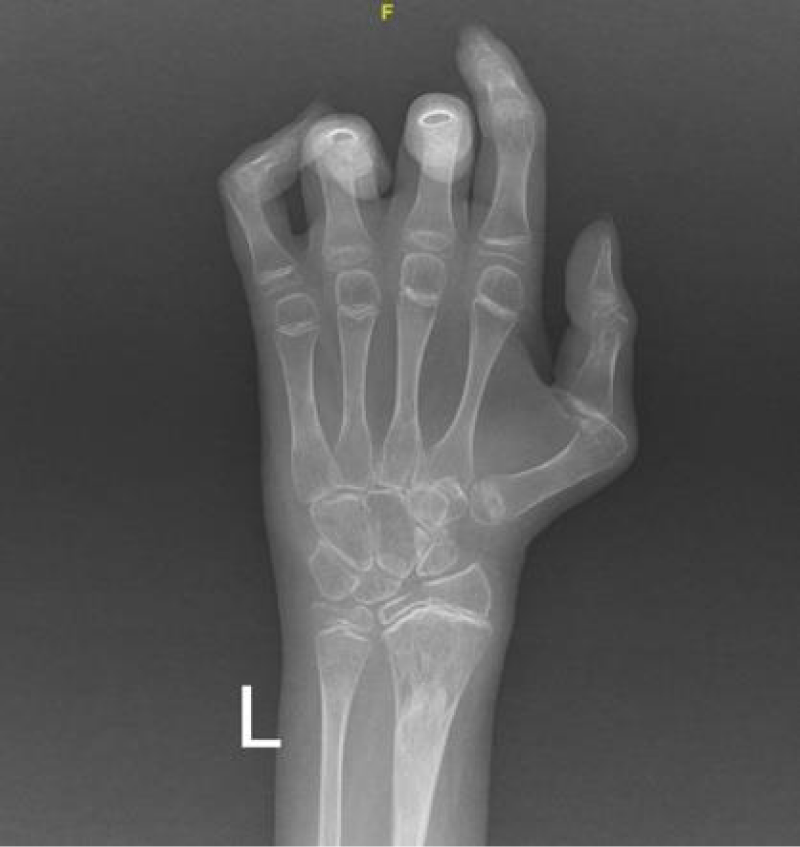
Figure 4: The X-ray of the left hand illustrated a generalized reduction in bone density with periarticular osteoporotic changes. Notably, a pathological fracture was identified in the distal left radius.
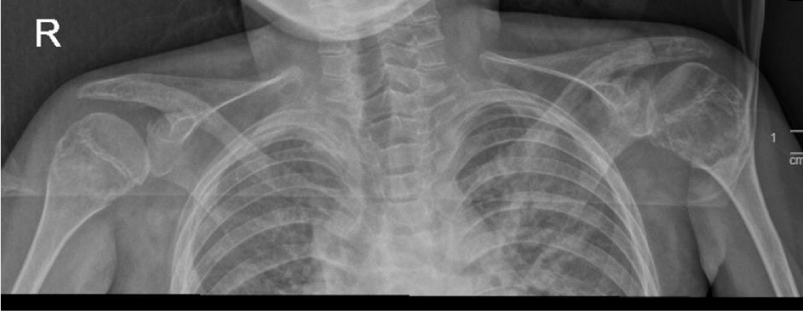
Figure 5: The chest X-ray showed degenerative changes identified in both shoulder joints. A lytic-like lesion was identified in the left humeral head, with a well-defined border and cortical expansion.
Electrocardiography showed resting tachycardia, with a heart rate of 160 bpm. Abdominal and pelvic ultrasounds showed bilateral grade three nephropathy. Additionally, abdominal and pelvic computed tomography without contrast showed bilateral irregular kidney contours of normal size without hydronephrosis. The scan also detected a Lamber-Bone lytic sclerotic lesion in the entire visualization portion of the lower dorsal spine, presenting with a compression fracture. Gibbus deformities in the L4–L5 vertebral bodies, as well as malalignment and displacement of the lumbar-dorsal spine, were evident (Figure 6), which corresponds with the findings of a Magnetic Resonance Imaging (MRI) performed in 2018.
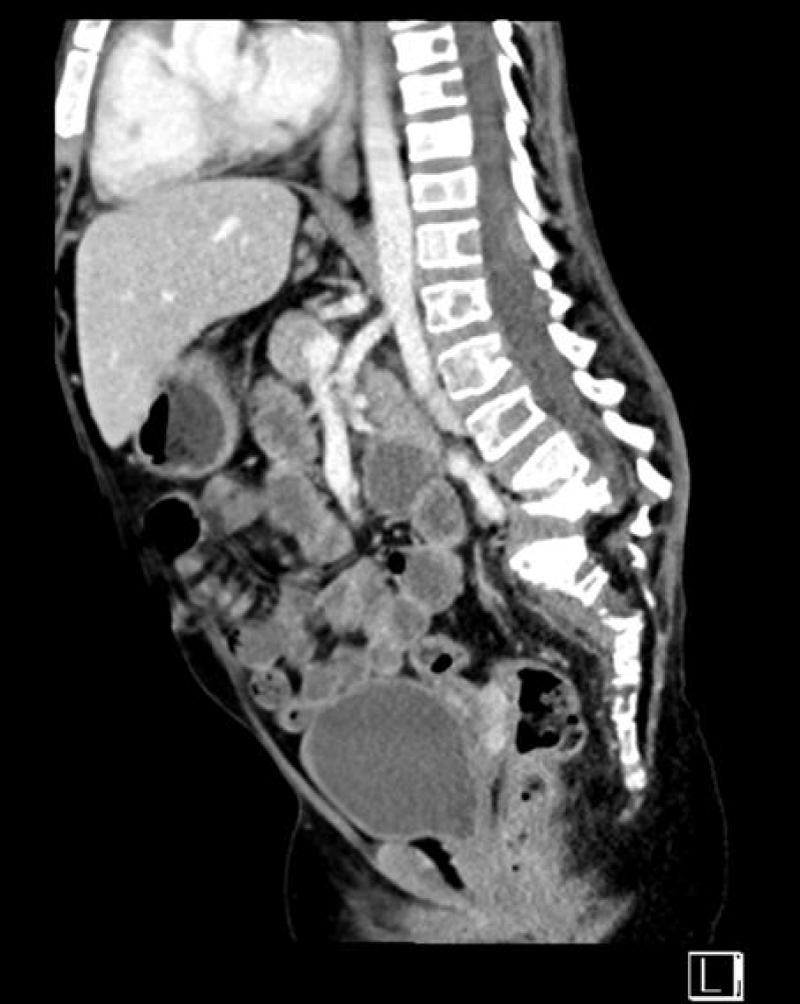
Figure 6: Abdominal and pelvic computed tomography without contrast revealed diffuse abnormal lytic and sclerotic bone lesions involving the entire visualized part of the spine, including the lower dorsal spine. Notably, it depicted a compression fracture or gibbus deformity of the lower vertebral bodies L4 and L5, along with malalignment and displacement of the lumbosacral spine.
A brain MRI conducted at our hospital showed a right-sided (25 x 9 mm) oblong-shaped cystic lesion displaying high signal intensity on T2-weighted and flair sequences and low-intensity T1, likely indicating an inflammatory mass ascending from the right ethmoidal air cells and extending to the right frontal sinus. Laboratory investigations revealed high inflammatory markers: an ESR (85 mm/hr) and CRP levels (7.7 mg/dl). Additionally, microcytic hypochromic anemia was observed, with a hemoglobin level of 8 g/dl. Genetic testing through Whole Exome Sequencing (WES) confirmed a positive result for the NOD2 [variant c.1442G>A p. (Gly481Asp)] gene mutation, consistent with autosomal dominant Blau syndrome. Amyloid protein levels were measured at 455 mg/dl (normal limit: up to 6.4 mg/dl). Serum Angiotensin-Converting Enzyme (ACE) levels were 52 U/L (normal range: 29–112 U/L). The tuberculin skin test results were negative. Tests for antinuclear antibody and rheumatoid factor were negative, while assays for complement components C3 and C4 remained within normal ranges. Two bone biopsies were conducted (first from the left radius and second from the left superior iliac crest one year later) due to bone pain, lytic lesions, and deformities. Both biopsies showed multiple non-necrotizing granulomatous lesions. Staining tests for acid-fast bacilli and periodic acid-Schiff were negative. Additionally, a skin biopsy showed multiple granulomatous lesions (Figure 7).
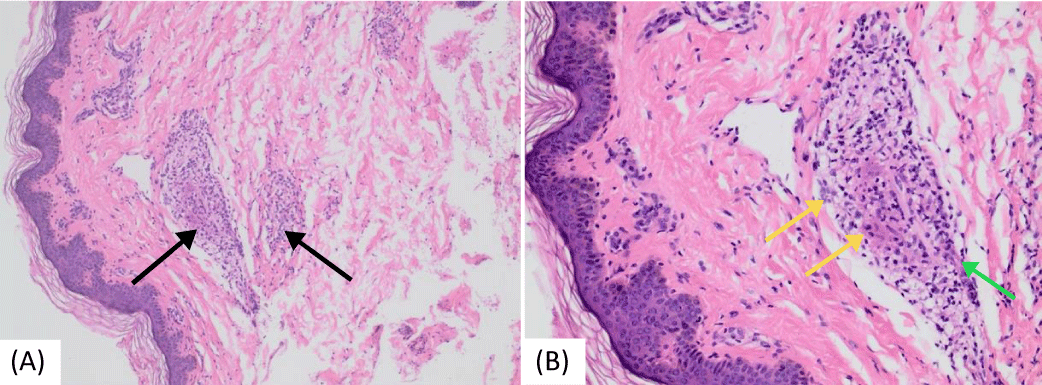
Figure 7: Histopathologic images of cutaneous sarcoidosis—a hematoxylin and eosin-stained section of skin from a punch biopsy taken from the back area. (a) Multiple non-caseating granulomas were found infiltrating the dermis and subcutis (black arrows) (b) Granulomas comprised epithelioid macrophages (yellow arrows) and a small number of lymphocytes (green arrow).
Accordingly, a diagnosis of early-onset sarcoidosis (EOS) was established. The patient received treatment with pulse steroids (30 mg/kg/day) being administered for three days, gradually tapering to a low dosage, in addition to Tocilizumab 162 mg given subcutaneously every 2 weeks. One month after starting treatment with Tocilizumab, the patient’s condition was showing marked improvement. Under the current management plan, the patient exhibited no subsequent disease progression, normalization of inflammatory markers, and a relatively stable clinical condition. Therefore, the steroid dosage was tapered to the lowest possible dose. Uveitis has been under control since treatment initiation, and she regularly follows up with ophthalmology.
Early-Onset Sarcoidosis (EOS) and Blau syndrome are both rare granulomatous autoinflammatory diseases affecting young children, caused by (the NOD2/CARD15) mutation. Affected children typically present with a triad of cutaneous manifestations, joint symptoms, and uveitis. Due to the variable clinical presentation of EOS, delays in diagnosis or misdiagnosis in EOS/Blau syndrome are not uncommon, particularly owing to the lack of awareness regarding these rare diseases. Accordingly, managing complications resulting from delayed diagnoses poses substantial challenges. Typically, early-onset sarcoidosis does not involve the lungs. However, a reported case in 2011 presented with intermittent episodes of fever, generalized rash, arthralgia, lymphadenopathy, and dyspnea. Chest X-ray findings revealed bilateral lower lung infiltration without hilar lymphadenopathy in this patient [8]. In 2016, a 7-year-old girl showed the characteristic triad of symptoms—rashes, arthritis, and uveitis—though she was initially misdiagnosed as having JIA. After an 8-week treatment with oral corticosteroids, there was minimal improvement observed in joint swelling and vision, while the rash showed no improvement. Routine laboratory results were within normal limits, and antibody test results were negative. Conversely, serum ACE was high, and both skin and synovial biopsy revealed multiple noncaseating granulomas. Consequently, a diagnosis of early-onset sarcoidosis was established for this case [9]. Similarly, in 2017, a 4-year-old female presented with a 2-year history of rash involving the face, chest, upper, and lower limbs, coupled with arthritis, uveitis, and massive right inguinal lymphadenopathy. Additional uncommon features, including onycholysis, nail ridging, and geographical tongue, were observed. A diagnosis of early-onset sarcoidosis was established based on her clinical presentation, raised serum ACE level, and histopathological evidence of noncaseating granulomas [10]. In 2019, Dr. Brian and his colleagues stated that the early presentation of arthritis in early-onset sarcoidosis often leads to the misdiagnosis of JIA in numerous cases [7]. An interesting case report in 2018 described an 11-year-old female with joint swelling and intermittent high fever, previously diagnosed as systemic JIA. However, subsequent genetic testing revealed a NOD2 mutation, ultimately categorizing her as a sporadic Blau syndrome case [11]. Another case report published in 2020 featured a 31-year-old Caucasian female recently diagnosed with Blau syndrome, who had previously been misdiagnosed as having systemic JIA since the age of 18 months [12].
Table 1 demonstrates a comparative analysis of our case with the other cases mentioned in the discussion, focusing on patient sex, clinical presentation, and age of onset.
| Table 1: Focusing on patient sex, clinical presentation, and age of onset. | |||
| Patient sex | Age of onset | Clinical presentation | |
| Our case | Female | 15 years old | Fever Joint pain Joint swelling Joint deformities Uveitis Skin lesions (papules) Contracture Lumbar kyphosis Short Status |
| Wong, et al. [8] | Female | 4 months old | Fever Joint pain Uveitis Skin lesions (papules) Lymphadenopathy Dyspnea |
| Argawal, et al. [9] | Female | 7 years old | Joint swelling Joint deformities Uveitis Skin lesions (papules) |
| Sahu, et al. [10] | Female | 2 years old | Joint swelling Uveitis Skin lesions (papules) Nail changes Lymphadenopathy |
| Imayoshi, et al. [11] | Female | 7 years old | Fever Joint swelling Uveitis |
| Ellis, et al. [12] | Female | 18 months old | Joint deformities Uveitis Contractures |
A scarce data exist on the epidemiology of pediatric sarcoidosis. A Danish National Registry study included 48 children within a cohort of 5536 patients with sarcoidosis, resulting in a calculated overall incidence for childhood sarcoidosis of 0.29 per 100,000 per year. The incidence ranged from 0.06 per 100,000 per year for children below 5 years old to 1.02 per 100,000 per year for children 14 to 15 years old [13-15]. An earlier international registry reported on 53 pediatric patients, of whom 14 had a family history, yielding a ratio of 1:5 for familial to sporadic forms. The International Registry of Pediatric Sarcoidosis, established in 2005, showed no gender difference or geographical predominance. It also stated that the majority of patients exhibiting the classic triad of arthritis, uveitis, and rash have disease onset before reaching 5 years old [16].
Skin lesions are the most commonly reported initial presentation in childhood early-onset sarcoidosis, observed in approximately 77% of patients with this condition [14]. The characteristic skin eruption associated with childhood early-onset sarcoidosis typically manifests as asymptomatic, fluctuating macular and papular rashes. These rashes tend to be symmetrical, and commonly located on the trunk and/or extremities [17]. Furthermore, other skin manifestations reported include hyperpigmented or hypopigmented lesions, ulcers, nodules, and subcutaneous tumors [2]. Consistent histopathology examination of these lesions demonstrates noncaseating granulomas with multinucleated giant cells. In our patient, the skin rash was described as multiple symmetrically distributed erythematous scaly papules on the trunks and bilateral upper and lower limbs, consistent with findings reported in other studies [18].
Joint manifestations usually present as symmetric polyarthritis, involving the interphalangeal joints, meta-carpophalangeal joints, wrists, feet, ankles, and occasionally the elbows. Contractures often appear early in the disease course, especially affecting the proximal interphalangeal joints, clinically described as camptodactyly [19]. Moreover, granulomatous inflammation in the periarticular structures frequently leads to marked periarticular swelling and the formation of tenosynovial cysts affecting the wrists and dorsa of the hands. These phenotypic observations exemplify characteristic features of the disease [20]. In our patient, multiple joint deformities were observed in the bilateral small joints of the hands and feet, accompanied by a notably restricted range of motion.
Uveitis is the most common ocular manifestation, affecting approximately 90% of patients diagnosed with early-onset childhood sarcoidosis [1]. It usually appears around the age of 12 as a late manifestation of the disease. The diagnosis of uveitis consistently poses challenges, with the identification of its etiology often proving to be a laborious task. The presence of concomitant conditions like Blau syndrome and atypical onset further adds complexity to the diagnostic process. Uveitis has also been reported in patients with JIA, and there is no definitive method to differentiate between uveitis occurring in JIA and childhood sarcoidosis. However, it is noteworthy that the antinuclear antibody test is positive in 88% of patients with JIA but typically negative in sarcoidosis [20]. This underscores the importance of collaborative efforts among pediatricians, rheumatologists, and ophthalmologists, emphasizing the necessity to enhance understanding of the disease. In fact, having bilateral uveitis is associated with an increased risk of visual morbidity. Thus, life-long follow-up and treatment for eye symptoms are essential for all patients. In the current hospital admission of our patient, active bilateral uveitis had been diagnosed two years prior to this recent admission. In the absence of a definitive blood test, the World Association of Sarcoidosis and Other Granulomatous Disorders (WASOG) recommends evaluating the serum ACE level. Typically, ACE levels are higher in children than in adults, and an increased ACE may contribute to the diagnostic process.
Diagnosis Blau syndrome primarily relies on clinical diagnosis, supported by histomorphology evidence of typical epithelioid noncaseating granuloma, after reasonably excluding other disorders known to cause granulomatous lesions. Alongside genetic testing, numerous studies have highlighted the involvement of NOD2/CARD15 in the beginning of granulomatous inflammation. In our patient, a WES test was conducted, revealing a mutation in the NOD2 gene, which substantiates our diagnosis.
In this report, we presented a unique case that illustrates the delayed presentation of early-onset sarcoidosis and the subsequent follow-up condition. Notably, our patient’s diagnosis of early-onset sarcoidosis was established after a 14-year duration. Unfortunately, she shows features of delayed diagnosis and complications associated with prolonged steroid usage, such as short stature and pathological spinal vertebrae fractures, which are devastating complications. The presentation of sarcoidosis varies considerably with age and often presents a diagnostic debate to clinicians due to the remitting and relapsing nature of the disease. Timely clinical and laboratory evaluation, along with imaging studies and histopathology findings, is imperative for the early diagnosis and treatment of sarcoidosis. This case indicates the importance of considering early-onset sarcoidosis in the differential diagnosis of pediatric patients presenting with arthritis and systemic manifestations such as fever, rash, and uveitis. Furthermore, a poor response to standard JIA therapy should prompt pediatricians to assess and consider such rare conditions. We acknowledge that delayed diagnosis emphasizes the potential for increased complications. This case serves as a compelling example, highlighting the urgent need for heightened awareness among physicians. Emphasizing the correlation between delayed diagnosis and heightened complications can significantly contribute to the ongoing dialogue about the importance of early detection and intervention in early-onset sarcoidosis.
Declarations
• Consent for publication: No direct identifiers are mentioned. Consent was taken from the Father.
• Availability of data and materials: Data “available on request”.
Authors’ contributions
MA was the primary physician for this patient and was a major contributor in writing and reviewing the manuscript. LB participated in following up with the patient and in writing the manuscript. SA carried out the dermatological examination and was involved in writing the manuscript. NA conducted the literature review and revised the manuscript. All the authors have read and approved the final manuscript.
- Shetty AK, Gedalia A. Sarcoidosis: a pediatric perspective. Clin Pediatr (Phila). 1998 Dec;37(12):707-17. doi: 10.1177/000992289803701201. PMID: 9864645.
- Shetty AK, Gedalia A. Childhood sarcoidosis: A rare but fascinating disorder. Pediatr Rheumatol Online J. 2008 Sep 23;6:16. doi: 10.1186/1546-0096-6-16. PMID: 18811966; PMCID: PMC2559831.
- Gedalia A, Khan TA, Shetty AK, Dimitriades VR, Espinoza LR. Childhood sarcoidosis: Louisiana experience. Clin Rheumatol. 2016 Jul;35(7):1879-84. doi: 10.1007/s10067-015-2870-9. Epub 2015 Jan 24. PMID: 25616361.
- Okafuji I, Nishikomori R, Kanazawa N, Kambe N, Fujisawa A, Yamazaki S, Saito M, Yoshioka T, Kawai T, Sakai H, Tanizaki H, Heike T, Miyachi Y, Nakahata T. Role of the NOD2 genotype in the clinical phenotype of Blau syndrome and early-onset sarcoidosis. Arthritis Rheum. 2009 Jan;60(1):242-50. doi: 10.1002/art.24134. PMID: 19116920.
- Rosé CD, Aróstegui JI, Martin TM, Espada G, Scalzi L, Yagüe J, Rosenbaum JT, Modesto C, Cristina Arnal M, Merino R, García-Consuegra J, Carballo Silva MA, Wouters CH. NOD2-associated pediatric granulomatous arthritis, an expanding phenotype: study of an international registry and a national cohort in Spain. Arthritis Rheum. 2009 Jun;60(6):1797-803. doi: 10.1002/art.24533. PMID: 19479837; PMCID: PMC2768567.
- White ES, Lynch JP 3rd. Current and emerging strategies for the management of sarcoidosis. Expert Opin Pharmacother. 2007 Jun;8(9):1293-311. doi: 10.1517/14656566.8.9.1293. PMID: 17563264.
- Chiu B, Chan J, Das S, Alshamma Z, Sergi C. Pediatric Sarcoidosis: A Review with Emphasis on Early Onset and High-Risk Sarcoidosis and Diagnostic Challenges. Diagnostics (Basel). 2019 Oct 25;9(4):160. doi: 10.3390/diagnostics9040160. PMID: 31731423; PMCID: PMC6963233.
- Wong LS, Ho JC, Yen YT. Early-onset childhood sarcoidosis: a case report. Dermatol Sin. 2011; 29.4: 125-128. doi:10.1016/j.dsi.2011.09.003.
- Agarwal K, Barua S, Adhicari P, Das S, Marak R. Early-onset sarcoidosis and juvenile idiopathic arthritis:A diagnostic dilemma. Indian J Dermatol Venereol Leprol. 2016 Sep-Oct;82(5):542-5. doi: 10.4103/0378-6323.183626. PMID: 27297270.
- Sahu P, Sharma S, Sharma N, Sharma S, Garg S. Unusual Clinical Presentations in Early-Onset Childhood Sarcoidosis: A Correlation or Coincidence? J Clin Diagn Res. 2017 Aug;11(8):WD01-WD03. doi: 10.7860/JCDR/2017/27841.10389. Epub 2017 Aug 1. PMID: 28969254; PMCID: PMC5620895.
- Imayoshi M, Ogata Y, Yamamoto S. A Case of Sporadic Blau Syndrome with an Uncommon Clinical Course. Case Rep Rheumatol. 2018 Dec 30;2018:6292308. doi: 10.1155/2018/6292308. PMID: 30693132; PMCID: PMC6332973.
- Ellis JC, Faber BG, Uri IF, J Emerson S. Early onset sarcoidosis (Blau syndrome): erosive and often misdiagnosed. Rheumatology (Oxford). 2020 May 1;59(5):1179-1180. doi: 10.1093/rheumatology/kez484. Erratum in: Rheumatology (Oxford). 2020 May 1;59(5):1190. PMID: 31620796.
- Byg KE, Milman N, Hansen S. Sarcoidosis in Denmark 1980-1994. A registry-based incidence study comprising 5536 patients. Sarcoidosis Vasc Diffuse Lung Dis. 2003 Mar;20(1):46-52. PMID: 12737280.
- Hoffmann AL, Milman N, Byg KE. Childhood sarcoidosis in Denmark 1979-1994: incidence, clinical features and laboratory results at presentation in 48 children. Acta Paediatr. 2004 Jan;93(1):30-6. PMID: 14989436.
- Petty, Ross E, Laxer, Ronald M, Lindsley, Carol B. Textbook of Pediatric Rheumatology E-Book (p. 2911). Elsevier Health Sciences. Kindle Edition.
- Lindsley CB, Petty RE. Overview and report on international registry of sarcoid arthritis in childhood. Curr Rheumatol Rep. 2000 Aug;2(4):343-8. doi: 10.1007/s11926-000-0073-z. PMID: 11123081.
- Schaffer JV, Chandra P, Keegan BR, Heller P, Shin HT. Widespread granulomatous dermatitis of infancy: an early sign of Blau syndrome. Arch Dermatol. 2007 Mar;143(3):386-91. doi: 10.1001/archderm.143.3.386. PMID: 17372104.
- Fernandez-Faith E, McDonnell J. Cutaneous sarcoidosis: differential diagnosis. Clin Dermatol. 2007 May-Jun;25(3):276-87. doi: 10.1016/j.clindermatol.2007.03.004. PMID: 17560305.
- Wouters CH, Maes A, Foley KP, Bertin J, Rose CD. Blau syndrome, the prototypic auto-inflammatory granulomatous disease. Pediatr Rheumatol Online J. 2014 Aug 6;12:33. doi: 10.1186/1546-0096-12-33. PMID: 25136265; PMCID: PMC4136643.
- Sfriso P, Caso F, Tognon S, Galozzi P, Gava A, Punzi L. Blau syndrome, clinical and genetic aspects. Autoimmun Rev. 2012 Nov;12(1):44-51. doi: 10.1016/j.autrev.2012.07.028. Epub 2012 Aug 2. PMID: 22884558.