
More Information
Submitted: April 30, 2024 | Approved: May 14, 2024 | Published: May 15, 2024
How to cite this article: Belcadi K, Isfaoun Z, El- Athmani O, El-Abdallaoui I, Ansari IN, et al. Juvenile Xanthogranulomatosis in a Hemophilic Boy: Case Report. J Adv Pediatr Child Health. 2024; 7: 062-065.
DOI: 10.29328/journal.japch.1001069
Copyright License: © 2024 Belcadi K, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Xanthogranulomatosis; Hemophilia; Histiocytosis

Juvenile Xanthogranulomatosis in a Hemophilic Boy: Case Report
K Belcadi*, Z Isfaoun, O EL-Athmani, I El-Abdallaoui, IN Ansari, M Lakhrissi, A Guindo, M El-Kababri, A Kili, L Hessissen, S Sefiani and M El-Khorassani
Hemophilia and Constitutional Hemorrhagic Disorders Treatment Center, Pediatric Hemato-Oncology Department, Rabat Children’s Hospital, Mohammed V University, Morocco
*Address for Correspondence: K Belcadi, Hemophilia and Constitutional Hemorrhagic Disorders Treatment Center, Pediatric Hemato-Oncology Department; Rabat Children’s Hospital, Mohammed V University, Morocco, Email: [email protected]
Juvenile Xanthogranulomatosis (JXG) is a condition, characterized by a proliferation of histiocytes, primarily observed in infants and young children. Cutaneous manifestations appear as yellow-orange-brown papules or nodules, typically localized on the face, neck, and upper chest. While most lesions regress spontaneously, some may require intervention for aesthetic, diagnostic, or hemorrhagic reasons.
A rare case of disseminated JXG in a child with hemophilia has been reported. In this patient with severe hemophilia A, cutaneous nodules appeared, some associated with bleeding requiring appropriate management. Treatment included the administration of factor VIII to prevent bleeding during surgical procedures and secondary prophylaxis, to control recurrent bleeding. The outcome was favorable with the disappearance of the cutaneous lesions without sequelae, under regular surveillance for both medical conditions.
This case highlights the rare association between juvenile xanthogranulomatosis (JXG) and hemophilia, a combination that has never been documented in the medical literature. This association only impacts the management of JXG when the cutaneous lesions bleed and their excision becomes necessary.
Juvenile Xanthogranulomatosis (JXG) results from an increased proliferation of histiocytes and represents the most common form of non-Langerhans histiocytoses. It typically affects infants and young children [1].
Cutaneous manifestations appear as papules or nodules, generally asymptomatic, with a yellow-orange-brown coloration mainly localized on the face, neck, and upper part of the torso [2]. While most lesions spontaneously regress within 3 to 6 years, some may persist and require excision for aesthetic, diagnostic, or hemorrhagic reasons before this period.
The association of JXG with hemophilia has never been described in the literature. In this regard, we report a case of JXG in a known hemophilic patient, with hemorrhagic complications requiring appropriate management. The objective of this sharing is to highlight management strategies associated with this unusual association and contribute to the enrichment of clinical knowledge and medical literature.
The case concerns a currently 12-year-old child, known to have hemophilia A with a factor VIII level of 0.25%. He has been followed in the Pediatric Hemato-Oncology Department since the age of 1 year and 4 months. At the age of 2 and a half, he progressively developed several well-defined, firm, rounded, brownish papules, less than 5 mm in diameter, sessile, non-follicular, with a smooth surface, localized in the external genitalia. Subsequently, subcutaneous nodules appeared on the scalp (Figure 1), thorax, and armpits, with the perception of well-defined, yellowish masses, one of which was complicated by bleeding.
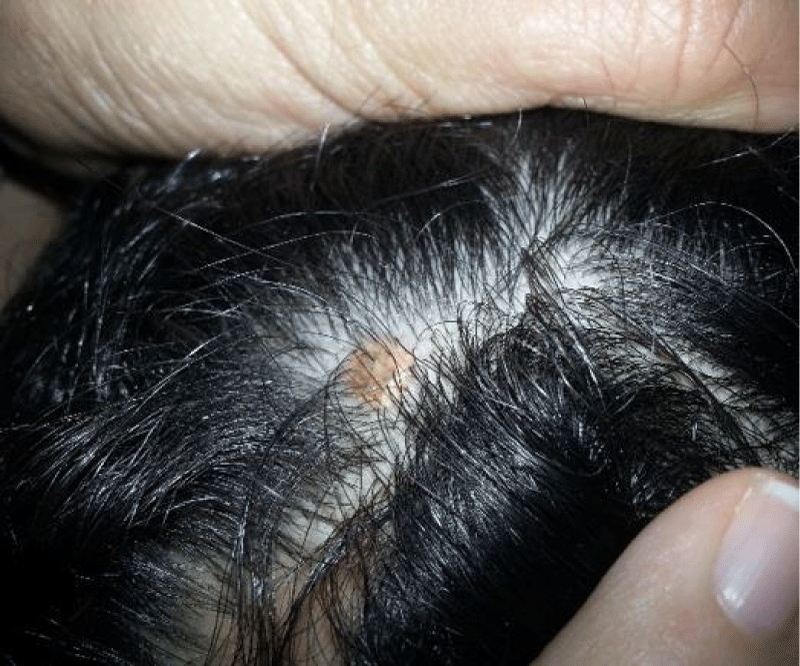
Figure 1: Cutaneous nodule on the scalp.
A biopsy-excision of two subcutaneous nodules on the scalp and a third one on the upper part of the right hemi-thorax was performed, after parental consent. We agreed on simultaneous circumcision. The procedure was scheduled with factor VIII coverage: prophylactic treatment, aiming to prevent any bleeding, consisted of administering 250 IU of factor VIII (20 IU/kg) before the procedure, with a second dose at H12 and a third one at H24.
The morphological study of the excised specimens suggested a cutaneous localization of histiocytosis (Figure 2).
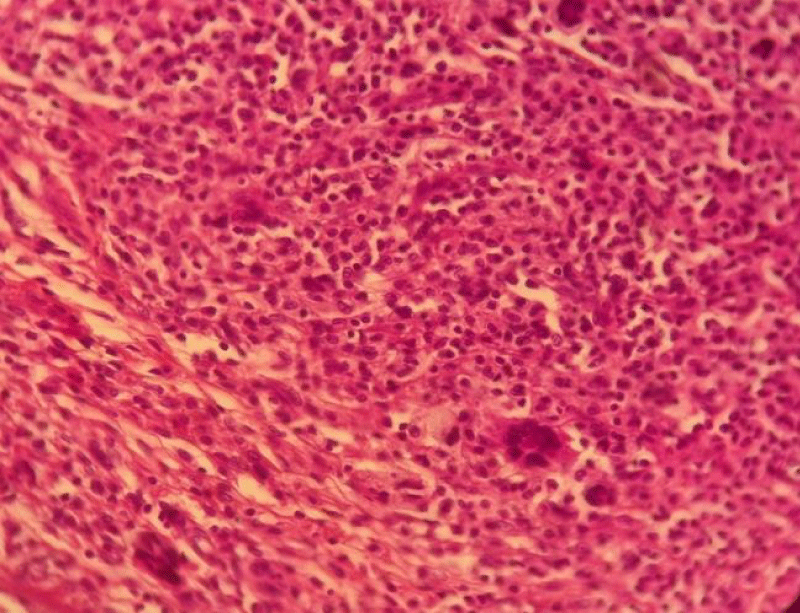
Figure 2: Standard histological examination (20x): the presence of a dermal infiltrate composed of monomorphic histiocytes, giant cells, and a polymorphic inflammatory infiltrate.
This was complemented by an immunohistochemical study which showed histiocytes negative for CD1a (Figure 3),positive for CD68 (Figure 4), and heterogeneously positive for S100, which supports a diagnosis of juvenile xanthogranulomatosis. The patient was discharged without experiencing any further bleeding.
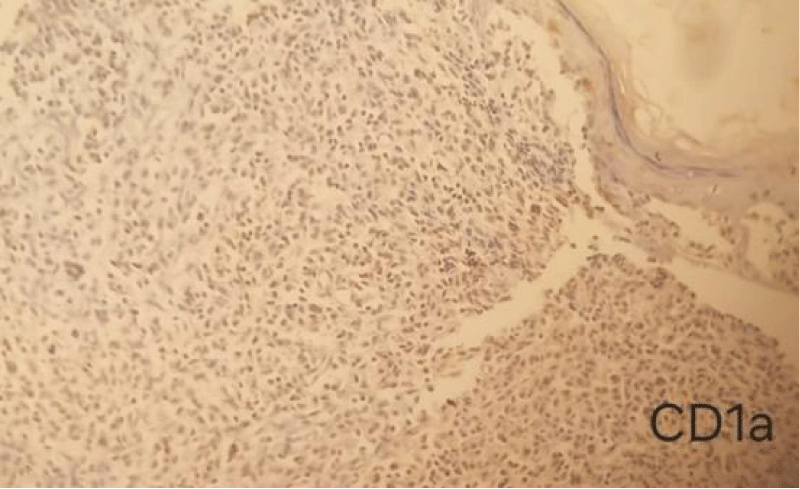
Figure 3: Immunohistochemistry of juvenile xanthogranuloma Negative for CD1a (IHC,10 x).
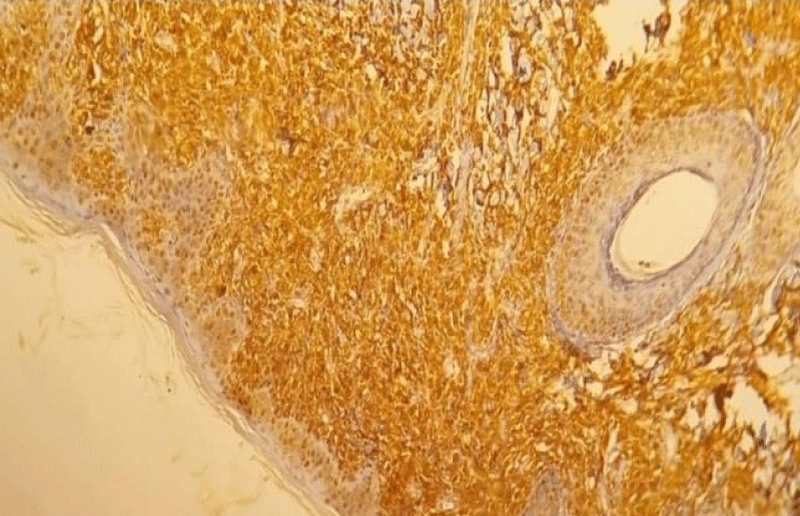
Figure 4: Immunohistochemistry of juvenile xanthogranuloma positive for CD68 (IHC,10 x).
Three months later, the child was consulted for active post-traumatic bleeding from a left axillary nodule. Surgical excision of the nodule was performed (Figure 5) under factor coverage at a dose of 20 IU/kg, followed by secondary prophylaxis with the administration of 20 IU/kg of factor three times per week as part of the minor surgery protocol. The outcome was favorable with healing after one month.
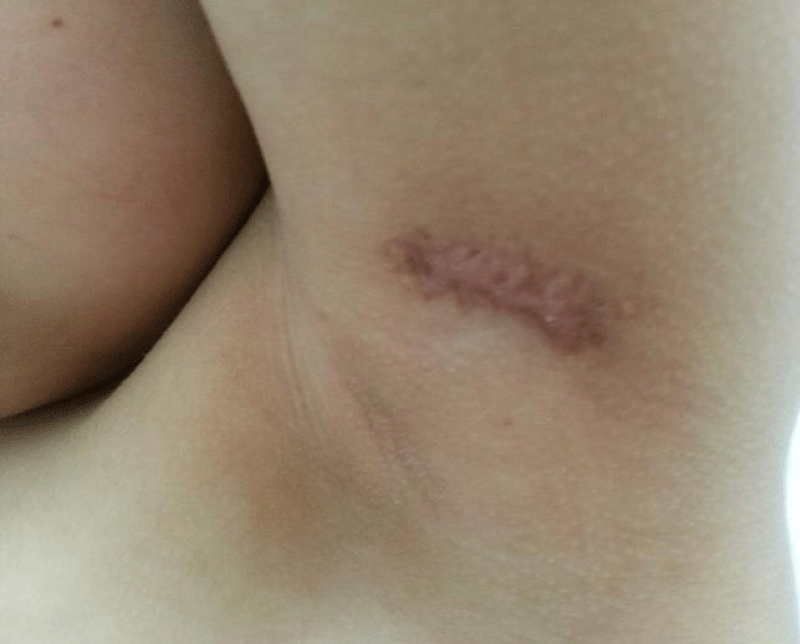
Figure 5: Excision of an axillary nodule.
Since then, he has not experienced any more bleeding, and the lesions have disappeared without leaving any sequels.
It is important to note that the prophylaxis implemented is secondary prophylaxis as part of his hemophilia management. Regular monitoring of his histiocytic pathology is conducted.
Currently, the child is 12 years old: with a 10-year follow-up, he is regularly monitored for his hemophilia. He has no remaining JXG nodules, and no other lesions have appeared since he was 2 years old.
Juvenile Xanthogranulomatosis (JXG) is a proliferative disease classified among non-Langerhans histiocytoses, primarily affecting infants and children [1].
It typically manifests as a single cutaneous involvement, although multiple forms have also been reported, including the case of our patient [3]. Clinically, typical manifestations are firm papules or nodules of yellow-orange-brown color, often affecting the neck, face, and upper chest. While oral lesions are rare, they often present as yellow nodules on the sides of the tongue, and can also appear on the gums, oral mucosa, and palate with the possibility of ulceration and bleeding [2].
Juvenile Xanthogranulomatosis (JXG) can also present with extracutaneous symptoms, affecting various organs such as muscles, lungs, liver, testicles, spleen, kidneys, and the central nervous system [1]. Ocular involvement is the most common extracutaneous manifestation, typically characterized by unilateral ocular xanthogranuloma, developing in about 0.5% of patients [2].
The classification of non-Langerhans histiocytoses is based on the presence of isolated cutaneous lesions or multiple diffuse cutaneous-mucosal lesions. Among the non-Langerhans histiocytoses with isolated cutaneous lesions, we find Juvenile Xanthogranulomatosis (JXG), which primarily affects children and often localizes on the upper body. This condition usually regresses spontaneously and is often characterized by fewer than 5 nodules. However, cases of JXG with more than 5 nodules have been reported, including our patient.
Xanthoma papules, affect adolescents and young adults, with a predominant localization on the trunk and extremities, presenting as a single nodule. Finally, reticulohistiocytoma, specific to young adults, can localize throughout the body and is characterized by a single small-sized nodule (< 5 mm).
The cells of JXG almost constantly express factor XIIIa. However, xanthoma papules and cutaneous reticulohistiocytoma, not expressing it, would rather be of macrophage origin. As for histiocytoses with diffuse cutaneous lesions, benign cephalic histiocytosis affects infants, presenting as papules and macules localized on the head and neck, and often regresses spontaneously. Generalized eruptive histiocytosis affects young adults, manifesting as reddish and symmetrical papules. Disseminated xanthoma, specific to young adults, presents as plaques and nodules, accompanied by general signs, and often localizes in skin folds. Multicentric reticulohistiocytosis, also affecting young adults, can affect all parts of the body and is accompanied by arthritis. Finally, progressive nodular histiocytosis, which can occur at any age, presents as large nodules, which can affect the head, limbs, and trunk, with a risk of disfigurement [3].
The definitive diagnosis of Juvenile Xanthogranuloma (JXG) relies on histopathological examinations. Classical histology of Juvenile Xanthogranuloma (JXG) reveals a dense, blade-shaped, non-encapsulated, and well-demarcated cellular infiltration in the dermis and upper part of the subcutaneous tissue. The cellular infiltrate includes five main types of cells (vacuolated, xanthomatous, spindle, scalloped, and oncocytic) ranging from monomorphic to mixed variants, with different types of multinucleated giant cells [4]. The microscopic appearance mainly depends on the duration of lesion evolution: early lesions show a monomorphic infiltrate of lipid-lacking macrophages that may occupy most of the dermis, while mature lesions contain abundant vacuolated foamy macrophages and multinucleated giant cells of Touton type, particularly at the dermal surface [4].
Touton cells are typical of Juvenile Xanthogranuloma (JXG) and are characterized by a ring of nuclei around lipid-rich cytoplasm. However, they are not observed in all cases and may be absent in up to 15% of patients. Although classically considered pathognomonic of JXG, Touton cells can also be present in other xanthomatous and histiocytic lesions (such as Erdheim-Chester disease and necrobiotic xanthogranuloma).
Immunohistochemical markers of JXG lesions bind to macrophage markers such as CD68, CD163, KiM1P, anti-FXIIIa, vimentin, and anti-CD4. They are generally negative for S-100, CD1a, and CD207 (Langerin), specific to Langerhans cells [4]. In the case presented, immunohistochemistry concluded with histiocytes positive for CD68 and negative for CD1a.
Generally, JXG lesions regress spontaneously in children, with spontaneous remission occurring in approximately 3 to 6 years, so they usually do not require treatment [5]. However, skin excisions may be performed at easily accessible sites in case of bleeding, aesthetic impairment, or ulceration.
The management of internal (non-cutaneous) lesions presents more significant challenges and may include various combinations of surgery 39,6%, chemotherapy 40,9%, radiotherapy 8,2%, and immunosuppression 2,5% [6]. Typically, chemotherapy and radiotherapy are considered for patients with symptomatic extracutaneous disease that is unresectable or partially resected [7]. For cases of ocular JXG, the use of topical or intralesional corticosteroids is common. In cases of rapid lesion progression or complications such as glaucoma or hyphema, systemic corticosteroids or surgery may be necessary.
The association of JXG with neurofibromatosis type 1 has been widely described [8], and a few rare cases of association with leukemias, notably juvenile chronic myelomonocytic leukemia, have been reported in the literature [9]. Due to this association, a detailed family history of NF1 and leukemia should be collected.
However, it seems that no cases of association with hemophilia have been reported, and no information was found during the search for articles regarding this association.
We report a case of JXG associated with hemophilia, which is not only a simple form of JXG but a disseminated form of late-onset, with some lesions complicated by bleeding. This is where the interest lies in reporting this unusual case, as managing such lesions is not easy, it must be standardized given the predisposition to bleeding and hemostatic delay in hemophilia patients. It should also be noted that there is probably no causal link between these cutaneous lesions and hemophilia.
This case underscores the rare association between juvenile xanthogranulomatosis (JXG) and hemophilia, a combination that has never been documented in the medical literature. This association only impacts the management of JXG when the cutaneous lesions bleed and excision becomes necessary. Surgery should be performed following the minor surgery protocol for hemophiliacs.
Ethical considerations
The informed consent of the patient as well as that of their parents for the publication of this case has been obtained.
- Ribeiro PH, Fritsch LN, Fernandes GJ, Oliveira AM, Ribeiro FB. Upper eyelid juvenile xanthogranuloma: a case report. Rev Bras Oftalmol. 2021; 80(5):e0030.
- Collie JS, Harper CD, Fillman EP. Juvenile Xanthogranuloma. 2023 Aug 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan–. PMID: 30252359.
- Emile JF, Charlotte F, Chassagne-Clement C, Copin MC, Fraitag S, Mokhtari K, Moreau A. Classification histologique et altérations moléculaires des histiocytoses [Histiocytoses: General classification and molecular criteria]. Presse Med. 2017 Jan;46(1):46-54. French. doi: 10.1016/j.lpm.2016.01.016. Epub 2016 Mar 4. PMID: 26952138.
- Hernández-San Martín MJ, Vargas-Mora P, Aranibar L. Juvenile Xanthogranuloma: An Entity With a Wide Clinical Spectrum. Actas Dermosifiliogr (Engl Ed). 2020 Nov;111(9):725-733. English, Spanish. doi: 10.1016/j.ad.2020.07.004. Epub 2020 Jul 25. PMID: 32721389.
- What Is a Juvenile Xanthogranuloma? Pediatr Dermatol. 2022 Jan;39(1):119-120. doi: 10.1111/pde.14920. PMID: 35106819.
- Zou T, Wei A, Ma H, Lian H, Liu Y, Wang D, Zhao Y, Cui L, Li Z, Zhang R, Wang T. Systemic juvenile xanthogranuloma: A systematic review. Pediatr Blood Cancer. 2023 May;70(5):e30232. doi: 10.1002/pbc.30232. Epub 2023 Feb 13. PMID: 36779547.
- Höck M, Zelger B, Schweigmann G, Brunner B, Zelger B, Kropshofer G, Kiechl-Kohlendorfer U. The various clinical spectra of juvenile xanthogranuloma: imaging for two case reports and review of the literature. BMC Pediatr. 2019 Apr 24;19(1):128. doi: 10.1186/s12887-019-1490-y. PMID: 31018833; PMCID: PMC6482499.
- Puerta P, Candela S, Rovira C, García Fructuoso G. Juvenile xanthogranuloma of skull base. Case report and review of the literature. Acta Neurochir (Wien). 2013 Jun;155(6):1039-40. doi: 10.1007/s00701-013-1718-9. Epub 2013 Apr 21. PMID: 23605373.
- Burgdorf WH, Zelger B. JXG, NF1, and JMML: alphabet soup or a clinical issue? Pediatr Dermatol. 2004 Mar-Apr;21(2):174-6. doi: 10.1111/j.0736-8046.2004.21219x. PMID: 15078363.